Microbial Source Tracking with 16S rRNA Sequencing: A Complete Guide for Biomedical Researchers
This comprehensive article explores the application of 16S rRNA gene sequencing for Microbial Source Tracking (MST) in biomedical and pharmaceutical contexts.
Microbial Source Tracking with 16S rRNA Sequencing: A Complete Guide for Biomedical Researchers
Abstract
This comprehensive article explores the application of 16S rRNA gene sequencing for Microbial Source Tracking (MST) in biomedical and pharmaceutical contexts. It begins by establishing the foundational principles of MST and the pivotal role of the 16S rRNA gene as a phylogenetic marker. The guide then details methodological workflows, from sample collection and primer selection to bioinformatic analysis and source attribution. A dedicated section addresses common pitfalls and optimization strategies to enhance accuracy and reproducibility. Finally, the article provides a critical evaluation of 16S rRNA sequencing against other MST techniques (e.g., qPCR, shotgun metagenomics) and discusses validation frameworks. Aimed at researchers, scientists, and drug development professionals, this resource synthesizes current best practices and future directions for leveraging microbial community data to ensure product safety and understand contamination pathways.
What is Microbial Source Tracking? The Foundational Role of 16S rRNA Gene Analysis
Article Content
Microbial Source Tracking (MST) refers to a suite of laboratory and computational methods used to identify the origins of microorganisms, particularly bacteria, in a given sample. In pharmaceutical and clinical settings, its primary objectives are to ensure product safety, maintain sterile manufacturing environments, diagnose infections, and prevent outbreaks. The advent of high-throughput 16S rRNA gene sequencing has revolutionized MST by providing a culture-independent, highly resolutive tool for microbial community profiling and source attribution.
Pharmaceutical Objectives:
- Contaminant Identification: Pinpoint the environmental or human source of microbial contaminants in non-sterile products, water-for-injection, or raw materials.
- Environmental Monitoring (EM): Track and differentiate resident from transient microbial populations in cleanrooms to validate cleaning procedures and identify breach points.
- Bioburden Control: Understand the microbial ecology of process water systems to implement targeted biocontrol strategies.
- Quality Assurance: Provide evidence for root-cause analysis during deviations and investigations, supporting regulatory submissions.
Clinical Objectives:
- Pathogen Source Attribution: Determine whether an infection is endogenous (e.g., gut translocation) or exogenous (e.g., hospital-acquired from a specific reservoir).
- Outbreak Investigation: Link clinical isolates from patients to specific environmental or point sources (e.g., contaminated equipment, plumbing) to halt transmission chains.
- Microbiome-based Diagnostics: Differentiate between colonization and infection by analyzing the source and dynamics of microbial communities in complex samples (e.g., respiratory, wound).
Integration with 16S rRNA Gene Sequencing: Within a thesis on 16S rRNA sequencing for MST, the technology serves as the core analytical engine. Sequencing of hypervariable regions generates operational taxonomic unit (OTU) or amplicon sequence variant (ASV) profiles. These profiles act as microbial "fingerprints" that can be compared against reference databases or source libraries using statistical or machine learning models (e.g., Bayesian classifiers, Random Forest) to probabilistically assign the sample to a likely source.
Table 1: Performance Metrics of Common MST Methods (Including 16S rRNA Sequencing)
| Method Category | Specific Method | Typical Resolution | Time-to-Result | Key Advantage | Primary Limitation |
|---|---|---|---|---|---|
| Library-Dependent | Ribotyping, BOX-PCR | Strain to Species | 3-5 days | High discriminatory power for cultured isolates | Requires isolate cultivation, limited library scope |
| Library-Independent | 16S rRNA Gene Sequencing | Genus to Species (Community-level) | 1-3 days | Culture-independent, comprehensive community profile | Limited resolution below genus/species for many taxa |
| Host-Specific Marker | PCR for Bacteroidales, Lachnospiraceae | Human vs. Animal Source | 1-2 days | Direct, specific, and rapid | May miss non-fecal contaminants, requires prior marker selection |
| Chemical Markers | Caffeine, Pharmaceuticals | Human/Urban Impact | Hours to days | Correlates with human activity | Not microbe-specific, subject to degradation |
Table 2: Example 16S Sequencing MST Study Outcomes in Clinical Settings
| Study Focus | Sequencing Platform | Key Finding (Quantitative) | Source Attribution Outcome |
|---|---|---|---|
| ICU Outbreak | Illumina MiSeq (V3-V4) | Patient and sink drain isolates shared >99.5% ASV similarity. | Confirmed hospital plumbing as persistent reservoir. |
| Catheter-Associated UTI | Ion Torrent PGM (V6-V8) | Urobiome of infected patients showed >30% similarity to gut microbiome profiles. | Supported endogenous gut origin as primary source. |
| Cleanroom Contamination | Illumina iSeq (V4) | Contaminant species comprised >85% of air sample community post-activity. | Traced to specific human activity during material transfer. |
Detailed Experimental Protocols
Protocol 1: 16S rRNA Gene Sequencing for MST from Environmental Swabs (Pharmaceutical Cleanroom)
Objective: To identify and track microbial sources via community analysis of cleanroom surface samples.
Materials: See "Research Reagent Solutions" below. Procedure:
- Sample Collection: Use sterile, DNA-free polyester swabs pre-moistened with sterile saline. Swab a defined area (e.g., 25 cm²) using a consistent template. Break swab head into a 2mL tube containing lysis buffer.
- DNA Extraction: Use a commercial kit optimized for low-biomass samples (e.g., DNeasy PowerSoil Pro Kit). Include negative control swabs and extraction blanks. Elute in 30-50 µL of elution buffer.
- 16S rRNA Gene Amplification: Perform PCR targeting the V3-V4 hypervariable region.
- Primers: 341F (5’-CCTACGGGNGGCWGCAG-3’) and 805R (5’-GACTACHVGGGTATCTAATCC-3’).
- PCR Mix: 12.5 µL 2x KAPA HiFi HotStart ReadyMix, 1 µL each primer (10 µM), 2 µL template DNA, 8.5 µL nuclease-free water.
- Cycling: 95°C 3 min; 25-30 cycles of [95°C 30s, 55°C 30s, 72°C 30s]; 72°C 5 min.
- Library Preparation & Sequencing: Index PCR, clean-up with magnetic beads, quantify, normalize, and pool. Sequence on an Illumina MiSeq with 2x300 bp paired-end chemistry.
- Bioinformatics & Source Tracking:
- Processing: Use QIIME2 or DADA2 for demultiplexing, quality filtering, denoising, chimera removal, and ASV clustering.
- Taxonomy: Assign taxonomy using a trained classifier (e.g., SILVA or Greengenes database).
- Analysis: Generate beta-diversity metrics (e.g., Bray-Curtis dissimilarity). Use Principal Coordinates Analysis (PCoA) to visualize clustering of samples by potential source (e.g., personnel, raw material airlock). Employ a source tracker algorithm (e.g., FEAST) to estimate proportional contributions from known source communities.
Protocol 2: Source Tracking for Clinical Infection Isolates
Objective: To compare clinical isolates to environmental isolates using 16S sequencing and phylogenetic analysis.
Procedure:
- Isolate Cultivation: Culture clinical (e.g., blood, urine) and suspected environmental (e.g., sink biofilm, ventilator surface) samples on appropriate media.
- DNA Extraction from Pure Cultures: Use a simple boiling prep or microbial DNA extraction kit.
- Full-Length 16S Gene PCR: Amplify near-full-length 16S rRNA gene.
- Primers: 27F (5’-AGAGTTTGATCMTGGCTCAG-3’) and 1492R (5’-GGTTACCTTGTTACGACTT-3’).
- Sequencing: Purify PCR product and sequence via Sanger sequencing.
- Analysis for MST:
- Sequence Alignment: Align clinical and environmental isolate sequences with reference sequences (e.g., from EzBioCloud database) using MAFFT.
- Phylogenetic Tree Construction: Build a maximum-likelihood tree (e.g., using MEGA software). Close clustering (high bootstrap value) of clinical and environmental sequences indicates a common source.
Mandatory Visualizations
Title: MST Workflow: From Sample to Source Attribution
Title: MST Method Selection Decision Tree
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for 16S rRNA-based MST Experiments
| Item / Reagent | Function / Purpose | Example Product / Specification |
|---|---|---|
| Low-Biomass DNA Extraction Kit | Optimized lysis and purification of microbial DNA from swabs, filters, or small volume samples while removing PCR inhibitors. | DNeasy PowerSoil Pro Kit (Qiagen), ZymoBIOMICS DNA Miniprep Kit. |
| High-Fidelity DNA Polymerase | Accurate amplification of the 16S rRNA gene target with minimal error rates for downstream sequencing fidelity. | KAPA HiFi HotStart ReadyMix, Q5 High-Fidelity DNA Polymerase. |
| 16S rRNA Gene Primers | Target-specific oligonucleotides for amplifying hypervariable regions (e.g., V4, V3-V4) or the near-full-length gene. | Illumina-adjusted 515F/806R (V4), 341F/805R (V3-V4), 27F/1492R (full-length). |
| Indexed Adapters & Library Prep Kit | For adding unique sample barcodes and Illumina/PacBio sequencing adapters to amplicons. | Nextera XT Index Kit, 16S Barcoding Kit (Oxford Nanopore). |
| Negative Control Material | Sterile water or swabs used to monitor and detect background contamination throughout the workflow. | DNA/RNA-Free Water, certified DNA-free swabs. |
| Mock Microbial Community | Genomic DNA from a defined mix of known bacterial strains. Serves as a positive control and for assessing pipeline accuracy. | ZymoBIOMICS Microbial Community Standard. |
| Bioinformatics Software | Tools for processing raw sequence data, taxonomic assignment, and statistical analysis for source comparison. | QIIME2, mothur, DADA2, FEAST (Fast Expectation-mAximization for microbial Source Tracking). |
Why the 16S rRNA Gene? Key Properties as a Universal Phylogenetic Marker
In the context of a thesis on Microbial Source Tracking (MST), the 16S rRNA gene serves as the foundational tool for profiling microbial communities to identify sources of fecal contamination in water, soil, and other environments. Its properties enable researchers to distinguish between human, agricultural, and wildlife fecal sources, which is critical for public health risk assessment and remediation strategies in drug development (e.g., for microbiome-based therapeutics) and environmental science.
Key Properties of the 16S rRNA Gene
The 16S ribosomal RNA gene is the standard chronometer for microbial phylogenetics and taxonomy due to a combination of essential properties.
Table 1: Key Properties of the 16S rRNA Gene as a Phylogenetic Marker
| Property | Description | Implication for MST/Phylogenetics |
|---|---|---|
| Ubiquitous Presence | Found in all prokaryotes (Bacteria and Archaea). | Allows for universal detection and comparison across all microbial life. |
| Functional Stability | Critical role in protein synthesis, constraining radical sequence change. | Sequence changes are largely due to evolution, not functional drift, making it a reliable historical record. |
| Appropriate Length | ~1,500 base pairs, containing both conserved and variable regions. | Provides enough information for robust analysis; conserved regions enable universal priming for PCR. |
| Variable Evolution Rates | Contains nine hypervariable regions (V1-V9) interspersed with conserved regions. | Hypervariable regions provide genus- or species-level discrimination; conserved regions allow for alignment across diverse taxa. |
| Low Horizontal Gene Transfer | Ribosomal RNA genes are rarely transferred horizontally between organisms. | Phylogeny reflects vertical inheritance and true evolutionary relationships, not recent gene exchange. |
| Large Reference Databases | Comprehensive databases (e.g., SILVA, RDP, Greengenes) contain millions of curated sequences. | Enables accurate taxonomic classification of newly sequenced amplicons, essential for source identification in MST. |
Application Notes for MST Research
Selection of Hypervariable Regions
The choice of hypervariable region for amplification significantly impacts taxonomic resolution in MST studies. Recent benchmarks indicate:
- V4-V5 region: Often provides the best balance between read length (with Illumina MiSeq) and taxonomic resolution for common gut bacteria.
- V1-V3/V3-V4 regions: Provide strong resolution for Bacteroidales, a key order for MST assays.
Table 2: Performance of Commonly Amplified 16S rRNA Gene Regions
| Region | Approx. Length (bp) | Key Strengths | Common MST Applications |
|---|---|---|---|
| V1-V3 | 500-600 | High resolution for many Bacteroides. | Human-specific source tracking. |
| V3-V4 | 450-500 | Broad phylogenetic coverage, standard for MiSeq. | General community profiling for source separation. |
| V4 | 250-300 | Excellent for short-read platforms, highly accurate. | High-throughput environmental screening. |
| V4-V5 | ~400 | Good resolution for Lachnospiraceae and Ruminococcaceae. | Discriminating between ruminant and other sources. |
| V6-V8 | 400-500 | Useful for specific phyla like Firmicutes. | Complementary region for validation. |
Quantitative Data from Recent Studies
Table 3: Example Quantitative Metrics from Recent 16S-based MST Studies
| Study Focus | Classifier Used | Accuracy/Resolution Reported | Key Insight for MST |
|---|---|---|---|
| Human vs. Non-human Source Discrimination | Random Forest on V4-V5 data | 95-99% Sensitivity/Specificity | Machine learning on 16S data can achieve high source prediction accuracy. |
| Geographic Variation of Gut Microbiota | Beta-diversity analysis (Weighted UniFrac) | Significant clustering (p<0.001, PERMANOVA) by host geography | Regional signatures must be accounted for in library-dependent methods. |
| Limit of Detection in Water Matrices | qPCR of host-associated 16S markers | 1-10 gene copies per reaction reliably detected | Sensitivity is sufficient for early contamination warning. |
Experimental Protocols
Protocol 1: Standard Workflow for 16S rRNA Gene Amplicon Sequencing in MST
Title: Comprehensive Workflow for 16S rRNA Gene Amplicon Sequencing in MST Research
Detailed Steps:
- Sample Collection & Preservation:
- Collect water (100-1000 mL filtered), soil (0.25-0.5 g), or fecal samples.
- Preserve immediately: freeze at -80°C, place in DNA/RNA shield, or use ethanol (for feces).
- DNA Extraction (Critical Step for Environmental Samples):
- Use a kit designed for environmental samples with inhibitors removal (e.g., DNeasy PowerSoil Pro Kit, MoBio).
- Include negative extraction controls.
- Quantify DNA using fluorescence-based assays (e.g., Qubit).
- PCR Amplification of 16S Hypervariable Region:
- Primers: Use barcoded versions of universal primers (e.g., 515F/806R for V4 region).
- Reaction: 25-50 µL volume. Use a high-fidelity polymerase (e.g., KAPA HiFi) to minimize errors.
- Cycling: Initial denaturation (95°C, 3 min); 25-35 cycles of [98°C 20s, 55°C 30s, 72°C 30s]; final extension (72°C, 5 min). Keep cycles low to reduce chimera formation.
- Include positive (mock community) and negative (no-template) PCR controls.
- Amplicon Cleanup & Library Pooling:
- Clean PCR products using magnetic bead-based purification (e.g., AMPure XP beads).
- Quantify cleaned amplicons, normalize concentrations, and pool equimolarly.
- Perform a final library QC (e.g., Bioanalyzer/TapeStation).
- Sequencing: Sequence on an Illumina MiSeq or iSeq platform using 2x250 bp or 2x300 bp chemistry to ensure overlap for paired-end assembly.
Protocol 2: Bioinformatic Analysis Pipeline using QIIME 2 (2024.2+)
Title: QIIME2 Pipeline for 16S Data Analysis
Detailed Steps:
- Import & Demultiplex: Use
qiime tools importandqiime demuxto generate a quality profile. - Denoising: Use
qiime dada2 denoise-paired(recommended) to correct errors, merge paired ends, remove chimeras, and generate Amplicon Sequence Variants (ASVs). - Taxonomic Assignment: Train a classifier on the latest SILVA or GTDB database for your specific primer region. Use
qiime feature-classifier classify-sklearn. - Phylogenetic Tree: Align ASVs with
qiime alignment mafft, mask positions, and build a tree withqiime phylogeny fasttreefor phylogenetic diversity metrics. - Diversity & Analysis:
- Rarefy feature table to even sampling depth:
qiime diversity core-metrics-phylogenetic. - For MST: Export feature table and taxonomy. Use SourceTracker2 (Bayesian approach) or machine learning classifiers (e.g., Random Forest in R) to estimate source contributions in sink samples.
- Rarefy feature table to even sampling depth:
The Scientist's Toolkit: Key Research Reagent Solutions
Table 4: Essential Materials and Reagents for 16S rRNA Gene-based MST
| Item Category | Specific Product Examples | Function in MST Workflow |
|---|---|---|
| DNA Extraction Kit | DNeasy PowerSoil Pro Kit (QIAGEN), FastDNA Spin Kit (MP Biomedicals). | Efficient lysis of diverse microbes and removal of potent environmental PCR inhibitors (humics, pigments). |
| High-Fidelity PCR Enzyme | KAPA HiFi HotStart ReadyMix (Roche), Q5 High-Fidelity DNA Polymerase (NEB). | Accurate amplification of the 16S target with minimal error rates, crucial for true ASV determination. |
| Universal 16S Primers | 515F/806R (V4), 27F/338R (V1-V2), 341F/785R (V3-V4). | Barcoded versions allow multiplexing. Select based on target taxa and sequencing platform. |
| Library Prep & Cleanup | AMPure XP Beads (Beckman Coulter), NEBNext Ultra II DNA Library Prep Kit. | Size selection and purification of amplicons, removal of primer dimers and contaminants. |
| Sequencing Standards | ZymoBIOMICS Microbial Community Standard (Zymo Research). | Mock community with known composition to validate entire wet-lab and bioinformatic pipeline accuracy. |
| Bioinformatic Databases | SILVA SSU Ref NR (v138.1+), RDP, GTDB. | Curated reference databases for accurate taxonomic classification of sequenced amplicons. |
| Analysis Software/Tools | QIIME 2, mothur, DADA2 (R), SourceTracker2, Phyloseq (R). | Processing raw sequences, statistical analysis, and specialized Bayesian source attribution modeling. |
Application Notes in 16S rRNA Gene Sequencing for MST Research
In Microbial Source Tracking (MST) research using 16S rRNA gene sequencing, the choice of sequence clustering or denoising method fundamentally shapes ecological interpretations and source attribution accuracy. These methodologies translate raw sequence data into biologically interpretable units.
OTUs are clusters of sequences, typically at a 97% similarity threshold, intended to approximate species-level groupings. This method reduces computational complexity and some sequencing error but can obscure true biological variation.
ASVs are resolved from denoising algorithms that infer exact biological sequences present in the sample, providing single-nucleotide resolution. This allows for reproducible, high-resolution tracking of microbial strains across studies.
Taxonomic Binning is the process of assigning these units (OTUs or ASVs) to taxonomic classifications using reference databases, enabling the biological identification crucial for MST.
The quantitative performance differences are summarized below.
Table 1: Comparative Analysis of OTU vs. ASV Methodologies for 16S rRNA-based MST
| Feature | OTU (97% clustering) | ASV (Denoising) |
|---|---|---|
| Resolution | Approximate (species-level) | Exact single-nucleotide |
| Repeatability | Variable; depends on clustering algorithm and parameters | High; reproducible across studies |
| Computational Demand | Lower | Higher |
| Error Handling | Clusters errors with true sequences | Attempts to model and remove sequencing errors |
| Sensitivity to Rare Taxa | May merge rare variants into abundant clusters | Better at distinguishing rare, true biological variants |
| Primary Tools | VSEARCH, USEARCH, mothur | DADA2, deblur, UNOISE3 |
| Ideal for MST when: | Budget/compute limited; broad source categories are sufficient | High-resolution tracking of specific host-associated strains is required |
Detailed Protocols
Protocol 1: DADA2 Pipeline for ASV Inference from 16S Paired-End Reads
Application: High-resolution profiling for discriminating closely related host sources.
- Filter and Trim: Use
filterAndTrim()in R. Trim forward reads to 240bp, reverse to 200bp. Truncate where quality drops below Q30. Remove reads with >2 expected errors. - Learn Error Rates: Model sequencing error rates (
learnErrors()) from a subset of data. - Dereplication: Combine identical reads (
derepFastq()). - Sample Inference: Apply the core denoising algorithm (
dada()) to infer true biological sequences. - Merge Paired Reads: Merge forward and reverse reads (
mergePairs()), requiring a minimum 12bp overlap. - Construct ASV Table: Make a sequence table (
makeSequenceTable()). - Remove Chimeras: Identify and remove bimera (
removeBimeraDenovo()). - Taxonomic Assignment: Assign taxonomy using
assignTaxonomy()against the SILVA reference database (v138.1 or newer).
Protocol 2: Closed-Reference OTU Clustering with VSEARCH for Rapid Analysis
Application: Standardized, database-dependent analysis for large-scale MST comparisons.
- Quality Control: Demultiplex and quality filter raw reads. Use
fastq_filterin VSEARCH (--fastq_maxee 1.0). - Dereplication: Dereplicate sequences (
--derep_fulllength). - Clustering: Cluster sequences at 97% similarity against a reference database (e.g., Greengenes 13_8) using
--usearch_globaland--id 0.97. - OTU Table Generation: Map all quality-filtered reads to the reference OTUs to create the final count table (
--otutabout). - Taxonomic Binning: Inherit taxonomy from the reference database used for clustering.
Protocol 3: Taxonomic Binning of ASVs/OTUs with QIIME 2 and a Custom Database
Application: Accurate source attribution using an MST-specific curated database.
- Data Import: Import the ASV/OTU representative sequences into QIIME 2 as a
FeatureData[Sequence]artifact. - Classifier Training: Train a naïve Bayes classifier (
q2-feature-classifier) on a custom MST 16S reference database (e.g., containing host-associated markers). - Taxonomic Assignment: Run the classifier on the ASV/OTU sequences.
- Confidence Thresholding: Apply a minimum confidence threshold (e.g., 0.7) to assignments. Unassigned features are labeled accordingly.
- Integration: Merge taxonomy results with the feature count table for downstream analysis.
Title: 16S rRNA Sequencing Analysis Workflow for MST
Title: From Community to Data: OTU & ASV Relationship
The Scientist's Toolkit: Research Reagent & Resource Solutions
Table 2: Essential Resources for 16S rRNA-based MST Analysis
| Item | Function in MST Research | Example Product/Resource |
|---|---|---|
| High-Fidelity DNA Polymerase | Minimizes PCR amplification bias and errors during library preparation, critical for ASV fidelity. | KAPA HiFi HotStart ReadyMix |
| 16S rRNA Primer Set (V3-V4) | Amplifies the target hypervariable region; choice influences taxonomic resolution and database compatibility. | 341F/806R (Earth Microbiome Project) |
| Mock Community (ZymoBIOMICS) | Validates entire wet-lab and computational pipeline, quantifying error rates and bias. | ZymoBIOMICS Microbial Community Standard |
| Positive Control DNA | MST-specific positive control (e.g., fecal DNA from target host) to confirm assay sensitivity. | Host-specific genomic DNA isolate |
| Silica-Bead Purification Kits | For consistent post-PCR clean-up and library normalization before sequencing. | AMPure XP beads |
| Reference Database | Curated collection of 16S sequences with taxonomy for binning; custom databases improve MST accuracy. | SILVA, Greengenes, custom MST database |
| Bioinformatics Pipeline | Containerized software for reproducible analysis (OTU/ASV, taxonomy, statistics). | QIIME 2, mothur, DADA2 R package |
| Computational Hardware | Sufficient RAM and multi-core CPUs for denoising algorithms and large-scale comparisons. | Minimum 16 GB RAM, 8+ cores recommended |
Within the framework of Microbial Source Tracking (MST) research using 16S rRNA gene sequencing, the identification of host-associated taxa is fundamental. This approach moves beyond quantifying fecal indicators to defining microbial signatures highly specific to a particular host source (e.g., human, cow, poultry). These signatures are composed of operational taxonomic units (OTUs) or Amplicon Sequence Variants (ASVs) that exhibit persistent and preferential association with one host species over others, often due to co-evolution and niche adaptation. Their application is critical for accurately attributing fecal pollution in environmental waters, assessing public health risks, and informing remediation strategies. For drug development, understanding host-specific gut microbiota can inform models for drug metabolism and toxicity studies. The core workflow involves: 1) Construction of a curated reference database from sequenced fecal samples of known origin, 2) Statistical identification of taxa with significant differential abundance across host groups, and 3) Validation of marker performance in blinded environmental samples.
Table 1: Common Host-Associated Microbial Markers in MST
| Host Source | Proposed Marker Taxa (Genus/Order) | Average Relative Abundance in Host (%) | Average Prevalence in Host Population (%) | Cross-Detection in Non-Target Hosts (%) |
|---|---|---|---|---|
| Human | Bacteroides (HF183, etc.) | 0.5 - 3.2 | >95 | <2 (ruminants, poultry) |
| Canine | Bacteroides (BacCan) | 0.1 - 1.5 | ~85 | <5 (human, avian) |
| Ruminant | Ruminococcaceae (Rum2Bac) | 0.01 - 0.5 | >90 | <1 (non-ruminants) |
| Avian | Helicobacter (Gull4) | 0.05 - 2.0 | ~70-80 | <10 (some mammals) |
Table 2: Performance Metrics of a Typical Marker Validation Study
| Metric | Human HF183 Assay | Ruminant Rum2Bac Assay |
|---|---|---|
| Sensitivity (True Positive Rate) | 96% | 92% |
| Specificity (True Negative Rate) | 99% | 98% |
| Limit of Detection (Gene Copies/PCR) | 10 | 25 |
| Environmental Sample Concordance | 89% | 85% |
Detailed Experimental Protocols
Protocol 1: Identification of Host-Associated Taxa from 16S rRNA Data Objective: To statistically identify taxa that are significantly enriched in one host source compared to others.
- Sample Collection & Sequencing: Collect fecal samples from ≥20 individuals per host source (e.g., human, cow, pig, chicken). Extract DNA using a validated kit (e.g., QIAamp PowerFecal Pro DNA Kit). Amplify the V4 region of the 16S rRNA gene using primers 515F/806R and sequence on an Illumina MiSeq platform (2x250 bp).
- Bioinformatic Processing: Process raw sequences using QIIME 2 (2024.5). Denoise with DADA2 to generate ASVs. Assign taxonomy using a pre-trained classifier (e.g., Silva 138.99) against the SILVA database. Rarefy the ASV table to an even sampling depth.
- Differential Abundance Analysis: Import the rarefied table into R. Use the
DESeq2orANCOM-BCpackage to identify ASVs differentially abundant between host groups. Apply a significance threshold of adjusted p-value (FDR) < 0.01 and a minimum log2 fold change > 2. - Marker Selection: Filter candidate ASVs for high prevalence (>80%) in the target host group and low prevalence (<5%) in non-target groups. Validate marker specificity in silico via BLAST against public databases.
Protocol 2: qPCR-Based Detection and Quantification of a Host-Associated Marker Objective: To quantify a specific host-associated genetic marker (e.g., HF183) in environmental water samples.
- Standard Curve Preparation: Clone the target marker sequence into a plasmid vector. Prepare a 10-fold serial dilution from 10⁸ to 10¹ gene copies/µL.
- Environmental DNA Extraction: Filter 100 mL of water through a 0.22 µm polycarbonate membrane. Extract DNA from the membrane using the DNeasy PowerWater Kit, eluting in 50 µL.
- qPCR Setup: Prepare reactions in triplicate containing: 10 µL of 2X TaqMan Environmental Master Mix, 0.9 µM of each primer (HF183F/HF183R), 0.25 µM TaqMan probe, 2 µL of template DNA (or standard), and nuclease-free water to 20 µL.
- Amplification: Run on a real-time PCR system with cycling: 95°C for 10 min; 45 cycles of 95°C for 15 sec and 60°C for 1 min (data acquisition).
- Analysis: Use the standard curve to interpolate gene copy numbers in samples. Apply any dilution factors to report copies per volume of original water sample.
Visualization Diagrams
Diagram 1: MST Workflow from HAT Discovery to Application
Diagram 2: Formation of Host-Associated Taxa
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for HAT Identification and Validation
| Item | Function & Application | Example Product |
|---|---|---|
| Fecal DNA Extraction Kit | Efficient lysis of tough microbial cells and inhibitors removal for reproducible metagenomic analysis. | QIAamp PowerFecal Pro DNA Kit |
| 16S rRNA Gene Primer Set | Amplifies hypervariable regions for taxonomic profiling. Widely adopted for consistency. | 515F/806R for V4 region |
| High-Fidelity PCR Master Mix | Accurate amplification for sequencing library preparation, minimizing errors. | KAPA HiFi HotStart ReadyMix |
| NGS Library Prep Kit | Prepares amplicons for Illumina sequencing with dual-index barcodes for multiplexing. | Illumina Nextera XT Index Kit |
| TaqMan Environmental Master Mix | Robust qPCR for inhibitor-prone environmental samples. Contains UNG to prevent carryover. | TaqMan Environmental Master Mix 2.0 |
| Cloning Vector Kit | Creates standard curves for absolute quantification in qPCR assays. | pCR4-TOPO TA Cloning Kit |
| Positive Control Plasmid | Contains target marker sequence for assay optimization and as run control. | Custom gBlock gene fragment cloned |
| Bioinformatics Pipeline | Integrated platform for 16S data processing, from raw reads to statistical analysis. | QIIME 2 (with DESeq2/ANCOM-BC plugins) |
Within the broader thesis on microbial source tracking (MST) using 16S rRNA gene sequencing, this document details application notes and protocols for three critical fields. These methods leverage high-resolution community profiling to identify, quantify, and track microbial contaminants, providing essential data for regulatory compliance, public health, and product safety.
Table 1: Summary of Key Application Areas and Associated Metrics
| Application Area | Primary Objective | Common Sequencing Metric (16S rRNA) | Typical Turnaround Time | Key Output |
|---|---|---|---|---|
| Contamination Investigation (Manufacturing) | Identify source of microbial deviation in sterile/non-sterile processes | Genus/Species-level identification; Community dissimilarity (Beta-diversity) | 3-7 days | Contaminant taxonomy report; Phylogenetic tree for source comparison. |
| Water Quality & Source Tracking | Determine fecal pollution sources (e.g., human, agricultural, wildlife) | Amplicon Sequence Variant (ASV) profiles; Host-associated genetic markers. | 5-10 days | Source contribution estimates; MST classification report. |
| Product Bioburden Analysis (Drug/Medical Device) | Characterize total viable microbial load on/in a product prior to sterilization. | Microbial load correlation with CFU; Biodiversity indices (e.g., Shannon Index). | 5-8 days | Bioburden identity and enumeration report; Risk assessment based on pathogen detection. |
Table 2: Representative Quantitative Outcomes from MST Studies Using 16S Sequencing
| Study Focus | Sample Type | Target Region | Key Quantitative Finding | Relevance to Application |
|---|---|---|---|---|
| Pharmaceutical Cleanroom Contamination | Air & Surface Swabs | V3-V4 | Staphylococcus and Micrococcus comprised >85% of contaminant flora. | Pinpoints human skin as primary contamination source, guiding sanitation protocols. |
| Urban Watershed Management | River Water | V4 | A single ASV from the genus Bacteroides of human origin accounted for 70% of the MST signal at the impaired site. | Accurately identifies wastewater leak, enabling targeted infrastructure repair. |
| Injectable Drug Product Bioburden | Pre-sterilization Bulk Solution | Full-length 16S | Detection of Ralstonia spp. at 0.1 CFU/mL, a level below traditional pharmacopoeial method thresholds. | Demonstrates superior sensitivity for risk mitigation regarding objectionable organisms. |
Experimental Protocols
Protocol 1: Comprehensive Workflow for Contamination Investigation via 16S rRNA Gene Sequencing
Objective: To trace the source of microbial contamination in a manufacturing environment.
- Sample Collection:
- Contaminated Material: Aseptically collect material (e.g., product, raw material, in-process sample). Include technical replicates.
- Potential Sources: Collect environmental samples (swabs from equipment, air filters, operator gloves, water) and reagent blanks.
- DNA Extraction (Critical Step):
- Use a kit optimized for low-biomass and inhibitory samples (e.g., with bead-beating for biofilms).
- Include extraction negative controls (lysis buffer only) to monitor reagent contamination.
- Library Preparation:
- Amplify the V3-V4 hypervariable region using primers 341F (5’-CCTAYGGGRBGCASCAG-3’) and 806R (5’-GGACTACNNGGGTATCTAAT-3’).
- Use a polymerase with high fidelity and low bias. Perform PCR in triplicate to mitigate stochastic effects.
- Include a PCR negative control (H₂O).
- Sequencing:
- Perform paired-end sequencing (2x250 bp or 2x300 bp) on an Illumina MiSeq or equivalent platform to achieve sufficient depth (~50,000 reads per sample).
- Bioinformatic & Statistical Analysis:
- Process reads through a pipeline (e.g., QIIME 2, DADA2) for quality filtering, denoising into ASVs, chimera removal, and taxonomy assignment against a curated database (e.g., SILVA, Greengenes).
- Conduct beta-diversity analysis (Principal Coordinates Analysis using Bray-Curtis dissimilarity). Statistically compare the contaminated sample community to potential source communities using PERMANOVA.
- Construct a phylogenetic tree of contaminant ASVs and closely related reference strains for high-resolution source tracking.
Protocol 2: Water Quality and Microbial Source Tracking Protocol
Objective: To identify and quantify fecal pollution sources in environmental water.
- Study Design & Sampling:
- Implement a stratified sampling design (impacted sites, potential source waters, upstream reference sites).
- Collect large-volume water samples (≥1L) and filter onto 0.22μm membrane filters immediately or preserve at -80°C.
- DNA Extraction & Library Prep:
- Extract DNA from filters using a soil/microbe-specific kit.
- Amplify the V4 region using dual-indexed primers (515F/806R) in a single-step PCR. Use a defined mock community as a positive control.
- Sequencing & In-silico Analysis:
- Sequence to a depth of ~100,000 reads/sample.
- Process sequences to the ASV level. Use a custom, locally-relevant MST classifier (e.g., a Random Forest classifier trained on fecal source libraries containing human, bovine, avian, etc., markers) to classify sequences.
- Quantification & Reporting:
- Calculate the relative abundance of source-specific genetic markers. Apply machine learning models to estimate proportional source contributions.
- Correlate MST data with traditional fecal indicator bacteria (FIB) counts (e.g., E. coli).
Protocol 3: Enhanced Bioburden Analysis for Medical Products
Objective: To characterize the taxonomic composition of viable microbial communities associated with a product.
- Sample Processing (Viability-Centric):
- Use culture-based enrichment: Incubate product samples in non-selective broth (e.g., TSB) to amplify viable microorganisms.
- After incubation, pellet cells and proceed to DNA extraction. Note: This captures only cultivable/proliferating organisms.
- DNA Extraction with Host/Inhibitor Removal:
- If analyzing products with human cells (e.g., cell therapies), include a step to lyse and degrade mammalian DNA (e.g., using selective nucleases).
- Broad-Range Amplification:
- Use primers targeting the V1-V3 or V4 region for optimal taxonomic resolution across diverse phyla. Include internal amplification controls to detect PCR inhibition.
- Sequencing & Risk Analysis:
- Sequence and generate ASV tables.
- Compare detected taxa against recognized lists of objectionable organisms (e.g., USP <1111>, <1115>). Calculate biodiversity metrics to assess community complexity.
Visualizations
Title: Contamination Investigation Workflow
Title: Water Quality MST Analysis Pathway
Title: Bioburden Risk Assessment Decision Tree
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for 16S rRNA-based MST Applications
| Item | Function & Rationale | Example Product/Kit |
|---|---|---|
| Low-Biomass DNA Extraction Kit | Maximizes yield from samples with sparse microbial cells while minimizing co-extraction of inhibitors common in environmental/clinical samples. | DNeasy PowerSoil Pro Kit (QIAGEN); MasterPure Complete DNA & RNA Purification Kit. |
| High-Fidelity PCR Polymerase | Reduces amplification bias and errors during 16S library construction, ensuring accurate representation of community structure. | Q5 High-Fidelity DNA Polymerase (NEB); KAPA HiFi HotStart ReadyMix. |
| Mock Microbial Community (Standard) | Serves as a positive control and calibrator for evaluating sequencing run performance, pipeline accuracy, and quantification bias. | ZymoBIOMICS Microbial Community Standard. |
| Indexed 16S rRNA Primers | Allows multiplexing of hundreds of samples in a single sequencing run by attaching unique barcode sequences to each sample's amplicons. | 16S Illumina Amplicon Primers (e.g., 341F/806R) with Nextera-style indices. |
| Bioinformatic Pipeline Software | Provides a reproducible, standardized suite of tools for processing raw sequencing data into an analyzable ASV/OTU table. | QIIME 2, mothur, DADA2 (R package). |
| Curated 16S Reference Database | Essential for assigning taxonomic names to sequence variants with up-to-date and accurate phylogenetic information. | SILVA, Greengenes, RDP. |
| MST Marker Database | A custom or public collection of host-associated 16S sequences (e.g., human, cow, pig gut microbiomes) used to train classification algorithms. | FEZ (Fecal Expert Zoo source database); locally constructed libraries. |
From Sample to Source: A Step-by-Step 16S rRNA MST Protocol
Study Design and Sample Collection Strategies for Robust Source Comparison
Abstract This document provides detailed application notes and protocols for the design of microbial source tracking (MST) studies using 16S rRNA gene sequencing. Within the broader thesis of applying high-throughput sequencing for MST, we outline critical considerations for study design, sample collection, and data generation to ensure robust, statistically sound comparisons between contamination sources. These protocols are designed to minimize bias and maximize the reproducibility of findings for environmental and pharmaceutical applications.
Core Principles of MST Study Design
A robust study design is foundational for attributing microbial signatures to specific sources. Key principles include:
- Hypothesis-Driven Sampling: Define clear target sources (e.g., human, bovine, industrial effluent) and sink samples (e.g., contaminated water, manufacturing surface).
- Replication: Biological and technical replication are non-negotiable. A minimum of n=5 replicates per source is recommended for initial biomarker discovery, with n>10 preferred for robust model building.
- Temporal and Spatial Pairing: Collect source and sink samples concurrently to account for diurnal and seasonal microbial flux. Spatial proximity between matched sources and sinks must be documented.
- Controls: Include negative controls (e.g., sterile water processed identically to samples) and positive controls (e.g., mock microbial communities) in every batch to assess contamination and sequencing performance.
Quantitative Sampling Strategy Framework
The following table summarizes a tiered sampling strategy based on study scope and resources.
Table 1: Tiered Sampling Strategy for MST Studies
| Study Tier | Primary Goal | Recommended Sources | Replicates per Source | Total Samples (Min) | Sequencing Depth per Sample |
|---|---|---|---|---|---|
| Pilot/Target Discovery | Identify potential source-discriminatory taxa. | 3-4 major suspected sources | 5-7 | 15-30 | 20,000 - 50,000 reads |
| Model Training | Build a classification model (e.g., Random Forest). | All known sources in catchment | 10-15 | 50-100 | 30,000 - 70,000 reads |
| Validation & Monitoring | Test model on blind samples; routine surveillance. | Focus on key sources & sinks | 5-10 (for new validation samples) | Variable | 20,000 - 50,000 reads |
Detailed Sample Collection & Preservation Protocol
Protocol 1: Water Sample Collection for 16S rRNA Gene Sequencing
Objective: To aseptically collect and preserve microbial biomass from water sources for downstream DNA extraction and sequencing.
Materials (The Scientist's Toolkit):
- Sterile, DNA-free Polypropylene Bottles (1L): For sample collection, minimizing exogenous DNA contamination.
- Peristaltic Pump or Sterile Syringe (60mL): For collecting water from specific depths or turbulent flows.
- Sterile In-line Filter Holder (0.22µm pore size, polyethersulfone membrane): To capture microbial biomass.
- DNA/RNA Shield or Lifeguard Solution: Commercial preservation buffer that immediately stabilizes nucleic acids at ambient temperature.
- Cryogenic Vials (2mL): For storing preserved filters or pellets.
- Cooler with Ice Packs or Dry Ice: For transport if preservative is not used immediately.
- Ethanol (70%) and Bleach (10% v/v): For decontaminating equipment between sampling sites.
- Field Data Sheet: For recording metadata (GPS, pH, temp, conductivity, time).
Procedure:
- Site Preparation: Decontaminate gloves and sampling equipment with 70% ethanol followed by 10% bleach rinse at the sampling site.
- Sample Collection: Using the pump or syringe, pass a measured volume of water (typically 100mL-1L, depending on turbidity) through the sterile 0.22µm filter. Record the volume filtered.
- Biomass Preservation:
- (Option A - In-field Stabilization): Aseptically transfer the filter to a cryogenic vial containing 1-2mL of DNA/RNA Shield. Vortex vigorously. Store at ambient temperature for transport.
- (Option B - Cold Transport): Place the filter in a dry, sterile cryovial and immediately store on dry ice or at -20°C. Transfer to -80°C within 6 hours.
- Controls: At each site, open a sterile bottle of preservation fluid, pour it through a filter, and process as a field negative control.
- Metadata: Complete the field data sheet for each sample. Unique sample IDs must link physical samples to metadata.
Experimental Workflow for Library Preparation
Protocol 2: 16S rRNA Gene Amplicon Library Preparation (V3-V4 Region)
Objective: To generate sequencing-ready libraries from extracted genomic DNA using a standardized, dual-indexing approach to minimize index hopping.
Materials:
- Extracted gDNA (concentration > 1 ng/µL): Quantified via fluorometry (e.g., Qubit).
- KAPA HiFi HotStart ReadyMix: High-fidelity polymerase for accurate amplification.
- Illumina 16S Metagenomic Sequencing Library Prep Protocol-Compatible Primers: e.g., 341F (5’-CCTACGGGNGGCWGCAG-3’) and 806R (5’-GGACTACHVGGGTWTCTAAT-3’) with overhang adapters.
- Illumina Nextera XT Index Kit v2 (Sets A & B): For dual-indexing of samples.
- Agencourt AMPure XP Beads: For post-amplification purification and size selection.
- Bioanalyzer or TapeStation System: For library quality control and fragment size verification.
Procedure:
- First-Stage PCR (Target Amplification):
- In a 25 µL reaction, combine: 12.5 µL KAPA HiFi Mix, 5 µL gDNA (1-10 ng), 1.25 µL each of the forward and reverse overhang primers (1 µM).
- Thermocycling: 95°C for 3 min; 25 cycles of: 95°C for 30s, 55°C for 30s, 72°C for 30s; final extension at 72°C for 5 min.
- Purification: Clean PCR products with 1X volume of AMPure XP beads. Elute in 25 µL of 10 mM Tris buffer.
- Second-Stage PCR (Indexing):
- In a 50 µL reaction, combine: 25 µL KAPA HiFi Mix, 5 µL purified first-stage product, 5 µL each of unique i5 and i7 index primers.
- Thermocycling: 95°C for 3 min; 8 cycles of: 95°C for 30s, 55°C for 30s, 72°C for 30s; final extension at 72°C for 5 min.
- Library Purification & QC: Clean indexed libraries with 1X AMPure XP beads. Assess concentration (via Qubit) and fragment size distribution (via Bioanalyzer, expecting a ~550-630 bp peak).
Visualization of Study Design and Workflow
Diagram Title: MST Study Design and Workflow Phases
Table 2: Essential Research Reagent Solutions for MST
| Reagent/Material | Function in MST Protocol |
|---|---|
| DNA/RNA Shield (Zymo Research) | Inactivates nucleases and stabilizes community DNA/RNA at room temperature, critical for field sampling. |
| PowerWater DNA Isolation Kit (QIAGEN) | Optimized for efficient lysis of diverse microorganisms captured on filters and removal of PCR inhibitors. |
| KAPA HiFi HotStart ReadyMix (Roche) | High-fidelity polymerase for minimal bias amplification of the 16S rRNA gene target. |
| Illumina Nextera XT Index Kit v2 | Provides unique dual indices for multiplexing hundreds of samples, reducing index-hopping errors. |
| Agencourt AMPure XP Beads (Beckman Coulter) | For consistent, size-selective purification of PCR amplicons and final libraries. |
| ZymoBIOMICS Microbial Community Standard | Defined mock community used as a positive control to assess sequencing accuracy and bioinformatic pipeline performance. |
| DNeasy PowerSoil Pro Kit (QIAGEN) | For complex solid samples (e.g., feces, soil) associated with source collection. |
| Qubit dsDNA HS Assay Kit (Thermo Fisher) | Fluorometric quantification of low-concentration DNA, more accurate for metagenomic samples than absorbance. |
Within microbial source tracking (MST) research utilizing 16S rRNA gene sequencing, the selection of primers targeting specific hypervariable regions (V1-V9) is a foundational and critical step. The choice directly influences taxonomic resolution, community profile accuracy, and the detection of bias. This application note details the considerations, comparative data, and protocols for informed primer selection.
Comparative Analysis of Primer Pairs
The following tables summarize key performance metrics for commonly used primer sets targeting different variable regions, based on current literature and empirical data.
Table 1: Primer Sequences and Target Regions
| Primer Pair Name | Forward Primer (5'->3') | Reverse Primer (5'->3') | Target Region(s) | Amplicon Length (~bp) |
|---|---|---|---|---|
| 27F / 338R | AGAGTTTGATCMTGGCTCAG | TGCTGCCTCCCGTAGGAGT | V1-V2 | ~310 |
| 341F / 534R | CCTACGGGNGGCWGCAG | ATTACCGCGGCTGCTGG | V3-V4 | ~210 |
| 515F / 806R | GTGYCAGCMGCCGCGGTAA | GGACTACNVGGGTWTCTAAT | V4 | ~290 |
| 799F / 1193R | AACMGGATTAGATACCCKG | ACGTCATCCCCACCTTCC | V5-V7 | ~390 |
| 967F / 1386R | CAACGCGAAGAACCTTACC | GTGTACAAGGCCCGGGAACG | V6-V8 | ~410 |
| 1389F / 1510R | TTGTACACACCGCCC | CCTTCYGCAGGTTCACCTAC | V9 | ~120 |
Table 2: Performance Characteristics in MST Context
| Target Region | Taxonomic Resolution | Gram Bias | Amplicon Size Suitability for Platform | Common Artifacts/Challenges |
|---|---|---|---|---|
| V1-V2 | High for Firmicutes, Bacteroidetes | Some bias against Actinobacteria | Good for short-read (e.g., MiSeq) | High sequence variability can challenge alignment. |
| V3-V4 | Good general resolution | Low | Excellent for short-read (e.g., MiSeq, iSeq) | Well-balanced, widely used benchmark. |
| V4 | Moderate to good | Very low | Excellent for most platforms | Shorter length may reduce species-level resolution. |
| V5-V7 | High for certain phyla | Can under-detect Bacteroidetes | Good for short-read | Potential for higher PCR bias. |
| V6-V8 | Good for environmental samples | Variable | Good for short-read | Chimera formation can be elevated. |
| V9 | Lower (conserved region) | Minimal | Best for highly degraded DNA | Limited discriminatory power for close relatives. |
Detailed Protocol: Primer Validation and Library Preparation
Protocol 1: In Silico Specificity and Coverage Check
Objective: To computationally evaluate primer pair performance against a current reference database.
Materials: Test primer sequences, SILVA or RDP database, software (e.g., TestPrime on SILVA, DECIPHER PrimerSearch).
Procedure:
- Obtain the latest version of the SILVA SSU Ref NR database in FASTA format.
- Use the TestPrime tool (integrated in the SILVA website) or the
PrimerSearchfunction in the DECIPHER R/Bioconductor package. - Input the exact forward and reverse primer sequences, allowing for degenerate base positions.
- Set parameters: Maximum number of mismatches = 1-2; Target domain = Bacteria and/or Archaea as required.
- Execute the analysis. Record the percentage of aligned sequences and any systematic mismatches for major taxonomic groups relevant to your MST sample matrix (e.g., fecal, aquatic).
Protocol 2: Empirical Testing with Mock Community
Objective: To assess amplification efficiency, bias, and resolution using a defined genomic mixture. Materials: ZymoBIOMICS Microbial Community Standard, selected primer pairs, high-fidelity PCR master mix, Qubit fluorometer, Bioanalyzer. Procedure:
- Extract DNA from the mock community standard using your standard extraction kit.
- Set up PCR reactions in triplicate for each primer pair:
- 25 µL reaction volume.
- 1X high-fidelity PCR buffer.
- 200 µM each dNTP.
- 0.5 µM each forward and reverse primer.
- 1 U high-fidelity DNA polymerase.
- 1 ng template DNA.
- PCR Cycling: 95°C for 3 min; 25-30 cycles of (95°C for 30s, Tm for 30s, 72°C for 45s/kb); 72°C for 5 min.
- Purify amplicons using a bead-based clean-up system (e.g., AMPure XP).
- Quantify purified amplicons using Qubit.
- Assess quality and size using a Bioanalyzer with a High Sensitivity DNA chip.
- Sequence on an appropriate platform (e.g., Illumina MiSeq, 2x250 bp for V3-V4).
- Analyze Data: Process sequences through a pipeline (e.g., QIIME 2, mothur). Compare observed relative abundances to the known composition of the mock community to calculate bias. Assess alpha and beta diversity metrics between technical replicates.
Visual Guide: Primer Selection Workflow for MST
Diagram Title: Primer Selection Decision Workflow
The Scientist's Toolkit: Essential Reagents & Materials
| Item | Function in Primer Selection & Validation | Example Product/Brand |
|---|---|---|
| High-Fidelity DNA Polymerase | Minimizes PCR errors and bias during amplicon generation for validation and library prep. | Phusion Hot Start Flex (Thermo), KAPA HiFi HotStart ReadyMix. |
| Quantitative DNA QC Kit | Accurately measures genomic DNA and amplicon concentration for normalization. | Qubit dsDNA HS Assay Kit. |
| Fragment Analyzer System | Precisely assesses amplicon size distribution and quality before sequencing. | Agilent Bioanalyzer HS DNA chip, Fragment Analyzer. |
| Bead-Based Purification Kit | Cleans up PCR products and normalizes pools for sequencing. | AMPure XP Beads, SPRIselect. |
| Defined Microbial Community Standard | Provides a known truth set for empirical validation of primer bias and efficiency. | ZymoBIOMICS Microbial Community Standard, ATCC Mock Microbiome Standard. |
| 16S rRNA Gene Reference Database | Enables in silico evaluation of primer coverage and specificity. | SILVA SSU Ref NR, RDP, Greengenes. |
| Primer Design & Analysis Software | Facilitates degenerate base design and computational testing. | DECIPHER (R), TestPrime (SILVA), primerBLAST (NCBI). |
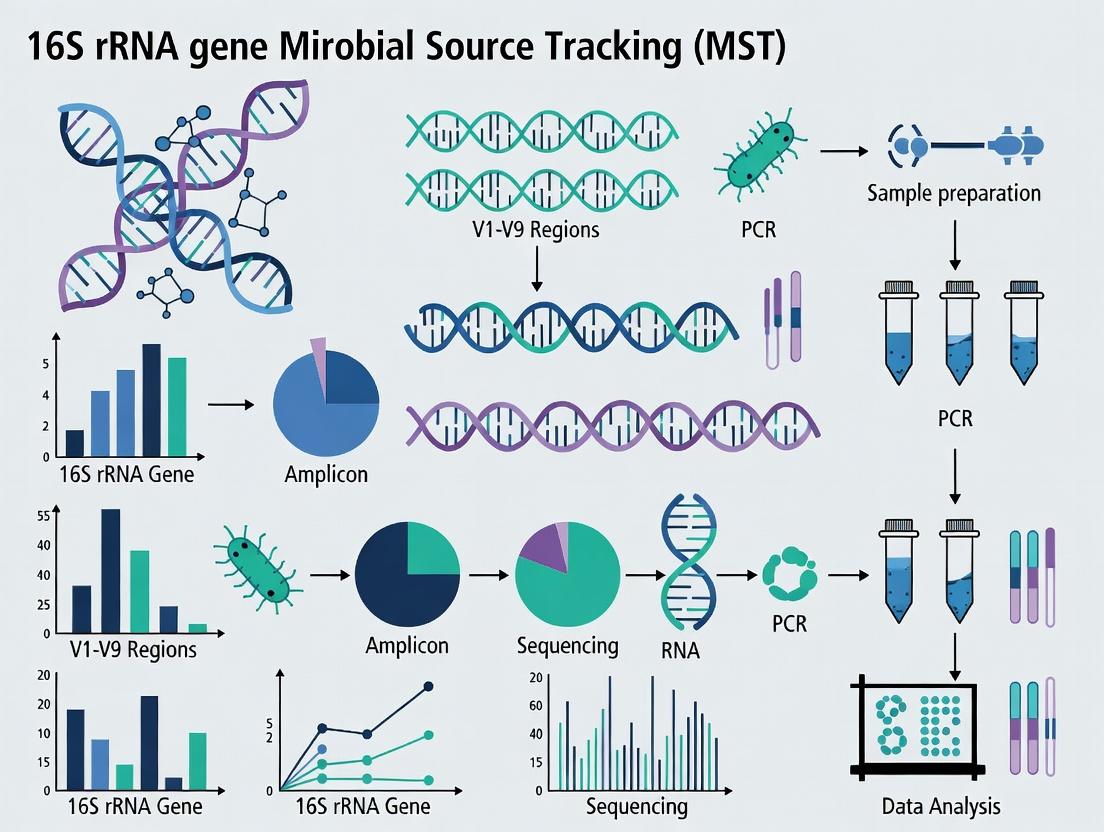
This protocol details a comprehensive wet-lab workflow for 16S rRNA gene sequencing within Microbial Source Tracking (MST) research. The process enables the characterization of microbial communities from complex environmental samples (e.g., water, soil) to identify fecal pollution sources. Standardization is critical for reproducibility and cross-study comparison.
Research Reagent Solutions and Essential Materials
| Item | Function in MST 16S rRNA Workflow |
|---|---|
| PowerSoil Pro Kit (Qiagen) | Inhibitor-removing DNA extraction kit optimized for environmental samples with tough-to-lyse cells. |
| PCR Primers (e.g., 515F/806R) | Target the V4 hypervariable region of the 16S rRNA gene for bacterial/archaeal profiling. |
| HotStart ReadyMix (KAPA) | High-fidelity, low-bias polymerase mix for accurate amplification of target regions. |
| Agencourt AMPure XP Beads | Solid-phase reversible immobilization (SPRI) beads for PCR product purification and size selection. |
| Nextera XT Index Kit (Illumina) | Provides unique dual indices and adapters for multiplexed library preparation compatible with Illumina sequencers. |
| Qubit dsDNA HS Assay Kit | Fluorometric quantification of double-stranded DNA with high sensitivity, critical for normalization. |
| Bioanalyzer High Sensitivity DNA Kit | Chip-based capillary electrophoresis for precise library fragment size distribution analysis. |
| Negative Extraction Control | Sterile water processed alongside samples to monitor contamination during DNA extraction. |
| Positive PCR Control (Genomic DNA) | Known genomic DNA (e.g., ZymoBIOMICS Microbial Community Standard) to assess PCR efficiency. |
Protocol 1: DNA Extraction from Environmental Samples
Objective: Obtain high-quality, inhibitor-free genomic DNA from filters or biomass for downstream PCR. Detailed Methodology:
- Sample Lysis: Transfer filter or up to 0.25 g of sample to a PowerBead Pro tube. Add solution CD1. Secure on a vortex adapter and vortex horizontally at maximum speed for 10 minutes.
- Inhibitor Removal: Centrifuge at 15,000 x g for 1 minute. Transfer supernatant to a clean tube. Add 200 µL of solution CD2, vortex for 5 seconds, incubate at 4°C for 5 minutes, then centrifuge at 15,000 x g for 3 minutes.
- DNA Binding: Transfer supernatant to a tube with 400 µL of solution CD3, vortex, and load onto an MB Spin Column. Centrifuge at 15,000 x g for 1 minute. Discard flow-through.
- Wash: Add 600 µL of solution EA (ethanol added) to the column. Centrifuge at 15,000 x g for 1 minute. Discard flow-through. Add 750 µL of solution AW1, centrifuge, discard flow-through. Add 750 µL of solution AW2, centrifuge for 3 minutes. Place column in a clean collection tube.
- Elution: Add 50-100 µL of nuclease-free water (preheated to 55°C) to the center of the membrane. Incubate for 5 minutes. Centrifuge at 15,000 x g for 2 minutes. Store DNA at -20°C. Quantitative QC Standards: DNA yield > 1 ng/µL, A260/A280 ratio of 1.8-2.0, A260/A230 > 1.7. Verify lack of inhibition via spike-in qPCR if needed.
Protocol 2: PCR Amplification of 16S rRNA V4 Region
Objective: Amplify the target hypervariable region with minimal bias and attach partial adapter sequences. Reaction Setup (50 µL):
| Component | Volume (µL) | Final Concentration/Amount |
|---|---|---|
| Genomic DNA (5 ng/µL) | 2 | 10 ng |
| Forward Primer (10 µM) | 2.5 | 0.5 µM |
| Reverse Primer (10 µM) | 2.5 | 0.5 µM |
| 2X HotStart ReadyMix | 25 | 1X |
| Nuclease-Free Water | 18 | - |
| Total Volume | 50 |
Thermocycling Conditions:
| Step | Temperature | Time | Cycles |
|---|---|---|---|
| Initial Denaturation | 95°C | 3 min | 1 |
| Denaturation | 95°C | 30 sec | |
| Annealing | 55°C | 30 sec | 25-30 |
| Extension | 72°C | 30 sec | |
| Final Extension | 72°C | 5 min | 1 |
| Hold | 4°C | ∞ |
Post-PCR Purification (SPRI Beads):
- Vortex AMPure XP beads thoroughly. Add 45 µL of beads (0.9X ratio) to 50 µL of PCR product. Mix thoroughly by pipetting.
- Incubate at room temperature for 5 minutes. Place on a magnetic stand for 2 minutes or until supernatant clears.
- Carefully remove and discard the supernatant.
- With tube on magnet, add 200 µL of fresh 80% ethanol. Incubate for 30 seconds, then remove and discard ethanol. Repeat wash. Air-dry beads for 5 minutes.
- Remove from magnet. Elute DNA in 33 µL of 10 mM Tris-HCl (pH 8.5). Mix, incubate for 2 minutes, place on magnet, and transfer 30 µL of purified eluent to a new tube.
- Quantify using Qubit. Expected yield: 10-50 ng/µL.
Protocol 3: Index PCR and Library Preparation
Objective: Attach full-length dual indices and Illumina sequencing adapters to purified amplicons. Index PCR Setup (50 µL):
| Component | Volume (µL) |
|---|---|
| Purified PCR Amplicon (5 ng/µL) | 5 |
| Nextera XT Index Primer 1 (N7xx) | 5 |
| Nextera XT Index Primer 2 (S5xx) | 5 |
| 2X HotStart ReadyMix | 25 |
| Nuclease-Free Water | 10 |
| Total Volume | 50 |
Thermocycling Conditions: Use the same cycle as Protocol 2, but reduce cycles to 8 to limit over-amplification. Library Cleanup & Normalization:
- Purify the Index PCR product using a 0.9X AMPure XP bead ratio (45 µL beads to 50 µL product) as in Protocol 2. Elute in 32.5 µL of Tris-HCl.
- Quantify all libraries using the Qubit dsDNA HS Assay.
- Pooling: Dilute each library to 4 nM based on Qubit concentration and average fragment size (~550 bp for V4 with adapters). Combine equal volumes of each 4 nM library into a final pool.
- Final QC: Assess the pooled library size distribution and molarity using the Bioanalyzer High Sensitivity DNA assay.
Table 1: Expected Yield and QC Metrics at Critical Stages
| Workflow Stage | Target Yield/Concentration | Key QC Metric & Target Value |
|---|---|---|
| Extracted DNA | >1 ng/µL (varies by sample) | Purity (A260/A280): 1.8-2.0 |
| Purified 1st PCR | 10-50 ng/µL | Fragment Size (Gel/TAE): ~400 bp (V4 insert) |
| Final Library Pool | 4 nM for sequencing | Fragment Size (Bioanalyzer): ~550 bp (with adapters) |
| Sequencing Loading | 6-20 pM (MiSeq v3) | Cluster Density: 800-1200 K/mm² |
Table 2: Common Troubleshooting Guide for MST 16S Workflow
| Problem | Possible Cause | Solution |
|---|---|---|
| Low DNA Yield | Inhibitors, inefficient lysis | Increase bead-beating time; use internal control. |
| No PCR Product | Inhibitors in DNA, primer mismatch | Dilute template; check primer specificity. |
| Smear on Gel | Over-amplification, primer dimers | Reduce PCR cycles; optimize annealing temperature. |
| Low Library Diversity | Over-dilution, poor bead cleanup | Accurate Qubit quantification; fresh AMPure beads. |
Workflow Diagrams
1. Introduction and Thesis Context
Within the broader thesis investigating Microbial Source Tracking (MST) using 16S rRNA gene sequencing, the choice of bioinformatic pipeline for processing raw sequence data is a critical determinant of result accuracy and ecological inference. This protocol details the application of three predominant pipelines—MOTHUR (a reference-based tool), DADA2 (a model-based approach), and QIIME 2 (a comprehensive, extensible platform)—in the context of MST research. Accurate delineation of host-specific microbial communities from environmental samples (e.g., water, soil) relies on precise amplicon sequence variant (ASV) or operational taxonomic unit (OTU) generation, demanding a rigorous and comparative understanding of these tools.
2. Comparative Summary of Pipelines
Table 1: Core Characteristics of DADA2, QIIME 2, and MOTHUR
| Feature | DADA2 | QIIME 2 | MOTHUR |
|---|---|---|---|
| Core Output | Amplicon Sequence Variants (ASVs) | ASVs or OTUs | Operational Taxonomic Units (OTUs) |
| Clustering Method | Model-based error correction; exact sequence inference. | Plugin-dependent (e.g., DADA2, deblur, VSEARCH). | Generally distance-based (e.g., 97% similarity). |
| Primary Approach | Error modeling and correction. | Modular, framework-based analysis. | Single, cohesive software package. |
| Primary Interface | R package. | Command line & graphical interface (Qiita). | Command line. |
| Key Strength | High-resolution, reproducible ASVs without clustering. | Extensive ecosystem, reproducibility, and visualization. | Mature, highly standardized SOPs, extensive reference alignment. |
| Typical Use in MST | High-resolution tracking of specific bacterial strains. | End-to-end analysis from raw data to statistical visualization. | Robust, traditional OTU-based community analysis. |
Table 2: Typical Quantitative Output Comparison (Theoretical Example from a Single 16S Dataset)
| Metric | DADA2 (ASVs) | QIIME 2 with DADA2 | MOTHUR (97% OTUs) |
|---|---|---|---|
| Input Reads | 1,000,000 | 1,000,000 | 1,000,000 |
| Post-Quality Filtered Reads | 850,000 | 850,000 | 830,000 |
| Non-Chimeric Reads | 800,000 | 800,000 | 790,000 |
| Final Features (ASVs/OTUs) | 2,150 | 2,150 | 1,850 |
| Singleton Features | ~120 | ~120 | ~350 |
| Computational Time (approx.) | Moderate | Moderate-High | High |
3. Experimental Protocols
Protocol 3.1: DADA2 Workflow for 16S rRNA Data (R Environment) Objective: Generate error-corrected ASVs from paired-end FASTQ files.
- Filter and Trim:
filterAndTrim(fwd, filt_fwd, rev, filt_rev, truncLen=c(240,200), maxN=0, maxEE=c(2,2), truncQ=2, compress=TRUE) - Learn Error Rates:
learnErrors(filt_fwd, multithread=TRUE)andlearnErrors(filt_rev, multithread=TRUE). - Dereplication:
derepFastq(filt_fwd)andderepFastq(filt_rev). - Sample Inference:
dada(derep_fwd, err=err_fwd)anddada(derep_rev, err=err_rev). - Merge Pairs:
mergePairs(dada_fwd, derep_fwd, dada_rev, derep_rev, minOverlap=12). - Construct Sequence Table:
makeSequenceTable(mergers). - Remove Chimeras:
removeBimeraDenovo(seqtab, method="consensus"). - Taxonomy Assignment: Use
assignTaxonomy(seqtab_nochim, "silva_nr99_v138.1_train_set.fa.gz")andaddSpecies().
Protocol 3.2: QIIME 2 Core Analysis via Command Line (using DADA2 plugin) Objective: Perform a complete analysis from raw data to diversity metrics.
- Import Data:
qiime tools import --type 'SampleData[PairedEndSequencesWithQuality]' --input-path manifest.csv --output-path demux.qza - Demultiplex and Summarize:
qiime demux summarize --i-data demux.qza --o-visualization demux.qzv - Denoise with DADA2:
qiime dada2 denoise-paired --i-demultiplexed-seqs demux.qza --p-trunc-len-f 240 --p-trunc-len-r 200 --p-trim-left-f 10 --p-trim-left-r 10 --o-table table.qza --o-representative-sequences rep-seqs.qza --o-denoising-stats stats.qza - Generate Taxonomy:
qiime feature-classifier classify-sklearn --i-classifier silva-138-99-nb-classifier.qza --i-reads rep-seqs.qza --o-classification taxonomy.qza - Create Phylogenetic Tree:
qiime phylogeny align-to-tree-mafft-fasttree --i-sequences rep-seqs.qza --o-alignment aligned-rep-seqs.qza --o-masked-alignment masked-aligned-rep-seqs.qza --o-tree unrooted-tree.qza --o-rooted-tree rooted-tree.qza - Diversity Analysis (Core Metrics):
qiime diversity core-metrics-phylogenetic --i-phylogeny rooted-tree.qza --i-table table.qza --p-sampling-depth 10000 --output-dir core-metrics-results
Protocol 3.3: MOTHUR Standard Operating Procedure (SOP) for MiSeq Data Objective: Generate 97% similarity OTUs following the established SOP.
- Make Contigs from Paired Ends:
make.contigs(file=stability.files) - Screen Sequences:
screen.seqs(fasta=current, group=current, maxambig=0, maxlength=275) - Filter Unique Sequences:
unique.seqs(fasta=current) - Align to Reference (e.g., SILVA):
align.seqs(fasta=current, reference=silva.v4.align) - Screen and Filter Alignment:
screen.seqs(fasta=current, count=current, start=your_start, end=your_end),filter.seqs(fasta=current, vertical=T, trump=.) - Pre-Cluster Sequences:
pre.cluster(fasta=current, count=current, diffs=2) - Chimera Detection (UCHIME):
chimera.uchime(fasta=current, count=current, dereplicate=t)andremove.seqs() - Classify Sequences:
classify.seqs(fasta=current, count=current, reference=trainset, taxonomy=trainset.tax) - Remove Non-Target Sequences:
remove.lineage(fasta=current, count=current, taxonomy=current, taxon='Chloroplast-Mitochondria-unknown-Archaea-Eukaryota') - Cluster into OTUs:
dist.seqs(fasta=current)followed bycluster(column=current, count=current) - Generate Shared File:
make.shared(list=current, count=current, label=0.03) - Taxonomy Summary:
classify.otu(list=current, count=current, taxonomy=current, label=0.03)
4. Visualized Workflows
DADA2 ASV Inference Workflow
QIIME 2 Modular Analysis Path
MOTHUR SOP for OTU Generation
5. The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Research Reagent Solutions for 16S rRNA Pipeline Analysis
| Item | Function in MST Pipeline Analysis |
|---|---|
| Silva or Greengenes Reference Database | Curated 16S rRNA sequence database for alignment, classification, and taxonomy assignment. |
| Naive Bayes Classifier (for QIIME2) | Pre-trained machine learning classifier (e.g., silva-138-99) for rapid taxonomic assignment. |
| Mock Community (ZymoBIOMICS, etc.) | Defined microbial mix used as a positive control to validate pipeline accuracy and error rates. |
| PCR Reagents & 16S Primer Set (e.g., 515F/806R) | For library preparation; targeting the V4 hypervariable region commonly used in MST studies. |
| MiSeq Reagent Kit v3 (600-cycle) | Standard chemistry for generating paired-end 300bp reads suitable for full 16S V4 coverage. |
| Qubit dsDNA HS Assay Kit | Fluorometric quantification of DNA concentration post-extraction and pre-amplification. |
| AMPure XP Beads | Magnetic beads for PCR product clean-up and size selection, removing primer dimers. |
| DNeasy PowerSoil Pro Kit | Standardized kit for efficient microbial genomic DNA extraction from complex environmental samples. |
| Positive Control Genomic DNA (e.g., E. coli) | Control for extraction and amplification efficiency. |
| Nuclease-free Water | Solvent for all molecular biology reactions to avoid RNase/DNase contamination. |
Microbial Source Tracking (MST) aims to identify the origins of fecal contamination in environmental waters. The use of 16S rRNA gene sequencing provides a high-resolution, culture-independent method to characterize microbial communities. A core challenge is translating complex community data into actionable source assignments. This necessitates the construction of robust, curated source libraries (known fecal samples from specific hosts) and the application of machine learning (ML) classifiers to interpret new, unknown samples against these libraries. These Application Notes detail the protocols and analytical frameworks for building 16S rRNA sequence-based source libraries and applying ML for classification, forming a critical methodology chapter for a thesis on advanced MST.
Protocol: Construction of a 16S rRNA Amplicon Sequence Variant (ASV) Source Library
Objective: To create a comprehensive, contamination-controlled, and biologically representative library of 16S rRNA gene profiles from known fecal sources.
Materials & Reagents:
- Sample Collection: Sterile spatulas, DNA/RNA Shield Fecal Collection Tubes (Zymo Research), dry ice or cold packs for transport.
- DNA Extraction: DNeasy PowerSoil Pro Kit (Qiagen) or equivalent, validated for low biomass and inhibitor removal.
- PCR Amplification: Primers targeting the V3-V4 hypervariable region (e.g., 341F/805R), high-fidelity DNA polymerase (e.g., Q5 Hot Start, NEB), PCR-grade water.
- Library Preparation & Sequencing: Illumina sequencing adapters and dual-index barcodes, AMPure XP beads (Beckman Coulter) for size selection, Qubit fluorometer for quantification. Sequences are generated on an Illumina MiSeq or NovaSeq platform using a 2x300 bp paired-end kit.
- Bioinformatics: Computational cluster or high-performance workstation, QIIME 2 (2024.5 or later), DADA2 plugin for denoising, SILVA v138.99 or Greengenes2 2022.10 database for taxonomic assignment.
Detailed Protocol:
- Strategic Sample Collection: Collect fresh fecal samples from target host groups (e.g., human, bovine, avian, swine). Include sufficient biological replicates (n≥50 per source) and geographic/temporal diversity. Include field blanks and extraction controls.
- Standardized DNA Extraction: Follow kit protocol with bead-beating step. Include negative extraction controls. Elute in 50 µL of elution buffer. Quantify using fluorometry; store at -20°C.
- Amplification & Barcoding: Perform triplicate 25 µL PCR reactions per sample using barcoded primers. Pool replicates. Run on gel to confirm amplicon size.
- Library Pooling & Sequencing: Normalize pooled amplicons using bead-based cleanup. Quantify final library pool by qPCR (KAPA Library Quant Kit). Sequence with appropriate PhiX spike-in (5-10%) for internal control.
- Bioinformatic Curation (Library Building):
- Demultiplexing & Quality Control: Import raw sequences into QIIME 2. Demultiplex based on barcodes, truncating reads based on quality plots (typically 280F/220R).
- Denoising & Chimera Removal: Apply DADA2 to infer exact Amplicon Sequence Variants (ASVs), removing chimeras de novo.
- Taxonomic Assignment: Assign taxonomy to ASVs using a pre-trained classifier on the reference database.
- Library Filtering: Remove ASVs present in negative controls. Filter out non-bacterial (mitochondrial, chloroplast) sequences. Apply a prevalence filter (e.g., retain ASVs present in >10% of samples within a source category) to reduce noise.
- Final Library Table: Produce a feature table (samples x ASVs), a taxonomy table, and a metadata file with source labels. This constitutes the source library.
Protocol: Machine Learning Classification of Unknown Samples
Objective: To train and validate a classifier model on the source library and apply it to classify unknown environmental samples.
Materials & Reagents:
- Software & Libraries: R (4.3.0+) with
tidymodels,caret,phyloseqpackages, or Python (3.10+) withscikit-learn,pandas,biom-format. Jupyter Notebook or RStudio for analysis. - Input Data: The curated source library (feature table, taxonomy, metadata) from Protocol 2.
Detailed Protocol:
- Data Preprocessing for ML:
- Normalization: Convert the ASV feature table to relative abundance (samples sum to 1).
- Feature Selection: Reduce dimensionality by filtering ASVs with low variance (e.g., variance < 0.001) or using phylogenetic-informed methods like
edgeR. - Train-Test Split: Partition the source library data into a training set (70-80%) and a held-out test set (20-30%), stratified by source label.
- Model Training & Validation (Using k-fold Cross-Validation on Training Set):
- Algorithm Selection: Test multiple algorithms: Random Forest (RF), Gradient Boosting Machines (XGBoost), and Regularized Logistic Regression (Lasso).
- Hyperparameter Tuning: Use grid or random search within a 10-fold cross-validation framework on the training set only to optimize parameters (e.g.,
mtryfor RF,learning_ratefor XGBoost). - Model Evaluation (CV Performance): Calculate cross-validated performance metrics (Accuracy, F1-Score, ROC-AUC).
- Final Model Evaluation & Interpretation:
- Test Set Assessment: Retrain the best model on the entire training set using optimal hyperparameters. Evaluate final performance on the untouched held-out test set. Report confusion matrix and metrics.
- Feature Importance: Extract and visualize the top 20-30 ASVs (features) contributing most to classification accuracy (e.g., via Mean Decrease in Gini for RF).
- Deployment for Unknown Sample Classification:
- Process unknown environmental samples through an identical wet-lab and bioinformatic pipeline (Protocol 2, steps 1-5).
- Projection: Normalize the unknown sample's ASV table identically to the training data.
- Prediction: Use the final, saved model to predict the source contribution probabilities for the unknown sample.
Data Presentation: Comparative Performance of ML Classifiers
Table 1: Cross-Validated Performance Metrics of ML Classifiers on a 16S rRNA Source Library
| Classifier | Average CV Accuracy (%) | Weighted F1-Score | ROC-AUC (Macro) | Key Advantage |
|---|---|---|---|---|
| Random Forest | 92.5 ± 3.1 | 0.921 | 0.989 | Robust to overfitting, handles non-linearities |
| XGBoost | 93.8 ± 2.8 | 0.932 | 0.991 | High predictive accuracy, feature importance |
| Lasso Regression | 88.2 ± 3.5 | 0.875 | 0.972 | Feature selection, interpretable coefficients |
| k-Nearest Neighbors | 85.7 ± 4.2 | 0.847 | 0.961 | Simple, no training phase |
Table 2: Final Test Set Performance of Optimized Random Forest Model
| Source Class | Precision | Recall | F1-Score | # Support (Samples) |
|---|---|---|---|---|
| Human | 0.95 | 0.91 | 0.93 | 45 |
| Bovine | 0.89 | 0.94 | 0.92 | 48 |
| Avian | 0.93 | 0.90 | 0.91 | 40 |
| Swine | 0.91 | 0.93 | 0.92 | 42 |
| Macro Avg | 0.92 | 0.92 | 0.92 | 175 |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents & Kits for 16S rRNA MST Library Construction
| Item | Supplier (Example) | Function in Workflow |
|---|---|---|
| DNA/RNA Shield Fecal Collection Tubes | Zymo Research | Preserves nucleic acid integrity at point of sample collection, inhibits microbial growth. |
| DNeasy PowerSoil Pro Kit | Qiagen | Standardized, high-yield DNA extraction with rigorous inhibitor removal for complex fecal samples. |
| Q5 Hot Start High-Fidelity DNA Polymerase | New England Biolabs | High-accuracy amplification of the 16S target region, minimizing PCR errors in library sequences. |
| Illumina MiSeq Reagent Kit v3 (600-cycle) | Illumina | Provides reagents for 2x300 bp paired-end sequencing, optimal for V3-V4 amplicon length. |
| Nextera XT Index Kit v2 | Illumina | Provides unique dual indices for multiplexing hundreds of samples in a single sequencing run. |
| KAPA Library Quantification Kit | Roche | Accurate qPCR-based quantification of final library pool for precise loading onto sequencer. |
Visualizations
Title: ML-Based MST Workflow from Sample to Prediction
Title: k-Fold Cross-Validation Model Training Process
Resolving Challenges: Optimization and Troubleshooting in 16S rRNA MST Studies
Within microbial source tracking (MST) research using 16S rRNA gene sequencing, achieving an accurate representation of microbial community structure is paramount. PCR amplification, a critical pre-sequencing step, introduces significant biases through primer-template mismatches and differential amplification efficiencies, compounded by excessive cycle numbers that distort relative abundances. This application note provides detailed protocols and data for mitigating these biases to enhance the fidelity of MST data.
Quantitative Data on Bias Effects
Table 1: Impact of Primer Mismatch and PCR Cycles on Community Representation
| Experimental Condition | Key Metric | Observed Effect | Reference |
|---|---|---|---|
| 338F/806R (V3-V4) vs. 27F/1492R (Full-length) | Shannon Diversity Index | 15-20% lower diversity in V3-V4 region vs. in silico full-length reconstruction. | (Klindworth et al., 2013) |
| Increased Primer Degeneracy (1 to 3 degenerate positions) | Amplification Efficiency Disparity | Up to 1000-fold difference in efficiency between template types. | (Bru et al., 2008) |
| PCR Cycles: 25 vs. 35 cycles | Ratio Deviation (Minor:Major Taxon) | 5- to 10-fold overestimation of minor taxa at 35 cycles. | (Kennedy et al., 2014) |
| Cycle Number Increase (25 to 40) | Coefficient of Variation (CV) for Abundant Taxa | CV increases from <5% to >25% for Bacteroidetes. | (Suzuki & Giovannoni, 1996) |
Experimental Protocols
Protocol 2.1: In Silico Primer Coverage and Mismatch Analysis
- Objective: Evaluate and select primer pairs for maximal coverage of target taxa with minimal mismatch.
- Procedure:
- Retrieve target 16S rRNA gene sequences from curated databases (e.g., SILVA, Greengenes) for expected taxa in your MST environment (e.g., fecal, aquatic).
- Align primer sequences to the aligned database using a tool like
search_oligosin mothur orTestPrimein SILVA. - Calculate the percentage of target sequences with perfect matches and with 1, 2, or >2 mismatches, particularly in the 3'-end region.
- Generate a coverage table by phylum/class. Select primers with >90% perfect match for your core taxa of interest.
Protocol 2.2: Empirical Testing of Primer Bias Using Mock Communities
- Objective: Quantify amplification bias introduced by candidate primer sets.
- Materials: Defined genomic DNA mock community (e.g., ZymoBIOMICS Microbial Community Standard).
- Procedure:
- Aliquot identical amounts of mock community DNA into separate PCR reactions for each primer set being evaluated (e.g., 515F/806R, 341F/785R).
- Perform PCR in triplicate under identical, low-cycle conditions (e.g., 25 cycles).
- Purify amplicons, index, and sequence on the intended platform (e.g., MiSeq).
- Bioinformatic Analysis: Process sequences (DADA2, QIIME2). Compare the observed proportions of each organism in the sequenced amplicons to the known genomic DNA proportions. Calculate a bias factor for each taxon:
Bias Factor = (Observed % / Known %).
Protocol 2.3: Determining the Optimal PCR Cycle Number
- Objective: Identify the minimum number of PCR cycles required for sufficient library yield without distorting community composition.
- Procedure:
- Set up a master mix from a single environmental DNA sample (e.g., water filtrate) and a mock community.
- Aliquot equal volumes into multiple tubes.
- Run identical PCR reactions but vary the cycle number (e.g., 20, 25, 28, 30, 35).
- Quantify yield (via fluorometry) for each cycle point.
- Sequence all reactions and analyze:
- Plot yield vs. cycles to identify the point where amplification exits exponential phase.
- For the mock community, calculate the deviation from expected composition at each cycle point using Bray-Curtis dissimilarity.
- For the environmental sample, monitor the stability of major taxa proportions across cycle numbers.
- Optimal Cycle: The highest cycle number before a significant increase in Bray-Curtis dissimilarity (e.g., >0.1) or major shift in dominant taxa.
Diagrams
Diagram 1: Workflow for Bias Mitigation in 16S MST
Diagram 2: PCR Cycle Impact on Community Fidelity
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Bias-Mitigated 16S Amplicon Sequencing
| Item | Function & Rationale |
|---|---|
| High-Fidelity DNA Polymerase (e.g., Phusion, Q5) | Reduces PCR errors and chimera formation due to superior proofreading activity, crucial for sequence accuracy. |
| Defined Mock Community (Genomic or Cell-based) | Provides a known truth standard for empirically quantifying primer and cycle bias during protocol optimization. |
| Low-Bias Primer Sets (e.g., 341F/785R, 515F/806R with parsimonious degeneracy) | Designed for broad coverage with minimal mismatches against target taxa, reducing amplification bias. |
| PCR Inhibitor Removal Kit (e.g., for humic acids in water) | Removes environmental inhibitors that cause differential amplification, a major source of bias in MST samples. |
| Fluorometric Quantification Kit (e.g., Qubit dsDNA HS Assay) | Accurately measures low DNA and amplicon concentrations without interference from RNA or salts, essential for cycle optimization. |
| Dual-Indexed Barcoded Adapters | Allows for unique, sample-specific indexing to prevent index hopping (crosstalk) and enable pooling of low-cycle PCR products. |
Addressing Low Biomass and Inhibitors in Environmental and Cleanroom Samples
In Microbial Source Tracking (MST) research utilizing 16S rRNA gene sequencing, sample integrity is paramount. The core thesis often hinges on accurately characterizing microbial communities to identify fecal pollution sources. However, environmental samples (e.g., water, soil) and ultra-clean environments (e.g., pharmaceutical cleanrooms) present two major, interconnected challenges: low microbial biomass and co-purified inhibitors. Low biomass increases susceptibility to contamination and stochastic variation in sequencing data, while inhibitors from humic substances, heavy metals, or cleaning agents can impede DNA extraction and downstream PCR amplification. Successfully overcoming these hurdles is critical for generating robust, reproducible data that can support valid inferences about microbial sources and community structures, forming a reliable foundation for the broader MST thesis.
Application Notes: Key Strategies and Comparative Data
Effective management of low-biomass, inhibitor-rich samples requires an integrated approach from collection to analysis. The following strategies are essential:
- Enhanced Biomass Recovery: Utilize filters with small pore sizes (e.g., 0.22µm) for water samples or high-efficiency swabs for surfaces. Sample concentration steps are non-negotiable.
- Inhibitor Removal: This often requires specialized lysis buffers and purification chemistries designed to sequester specific inhibitory compounds. Silica-membrane columns may be insufficient; alternative chemistries like chitosan, polyvinylpolypyrrolidone (PVPP), or enhanced wash buffers are often employed.
- PCR Optimization: The use of inhibitor-tolerant polymerases and adjuncts like bovine serum albumin (BSA) or betaine is standard. Increasing PCR cycle numbers for low biomass samples must be balanced against increased contamination risk and amplification bias.
- Rigorous Contamination Controls: Including negative controls (extraction blanks, PCR no-template controls) at every batch is mandatory to distinguish signal from noise. Reagent-only controls should be sequenced alongside samples.
Table 1: Comparison of Commercially Available DNA Extraction Kits for Challenging Samples
| Kit Name (Example) | Core Technology / Chemistry | Recommended for Inhibitor Type | Elution Volume (Typical) | Key Advantage for Low Biomass |
|---|---|---|---|---|
| DNeasy PowerSoil Pro Kit | Silica membrane + specialized inhibitor removal solution | Humic acids, phenols, polysaccharides | 50-100 µl | Optimized for soil; high inhibitor removal efficiency. |
| ZymoBIOMICS DNA Miniprep Kit | Bead beating + inhibitor removal technology | Humics, proteins, salts | 50-100 µl | Includes a DNase step to remove contaminating DNA. |
| Molzym MolYsis Basic | Selective host cell lysis + enzymatic degradation | Eukaryotic cell/human DNA background | 50 µl | Selectively enriches prokaryotic DNA, reducing host background. |
| Promega DNA IQ System | Paramagnetic resin | Broad spectrum, including some dyes | 50-100 µl | Scalable binding; efficient from swabs and filters. |
| Qiagen DNeasy Blood & Tissue (with pre-treatment) | Silica membrane | Proteins, salts | 100-200 µl | Flexibility for pre-lysis enzymatic or mechanical treatments. |
Table 2: PCR Adjuncts and Their Functions in Mitigating Inhibition
| Adjunct | Typical Working Concentration | Proposed Mechanism of Action | Common Use Case |
|---|---|---|---|
| Bovine Serum Albumin (BSA) | 0.1 - 1.0 µg/µL | Binds to inhibitors, sequestering them from Taq polymerase. | Humic/fulvic acids, polyphenols, heparin. |
| Betaine | 0.5 - 1.5 M | Reduces secondary structure in GC-rich templates; can enhance primer annealing. | High GC-content genomes, some ionic inhibitors. |
| Tween-20 | 0.1 - 1.0% | Non-ionic detergent that can disrupt inhibitor-enzyme interactions. | Non-specific protein binding. |
| Polyvinylpyrrolidone (PVP) | 0.1 - 1.0% | Binds polyphenolic compounds through hydrogen bonding. | Plant-derived polyphenols, tannins. |
Experimental Protocols
Protocol 1: Concentrated Filtration and Extraction from Low-Biomass Water Samples
Objective: To concentrate microbial cells from large-volume water samples and extract inhibitor-free DNA suitable for 16S rRNA gene PCR. Materials: Peristaltic pump, filtration manifold, 0.22µm mixed cellulose ester filters, sterile forceps, DNA extraction kit (e.g., DNeasy PowerWater Kit or equivalent), sterile scissors, 2ml bead-beating tubes.
Procedure:
- Filtration: Aseptically place a sterile 0.22µm filter on the filtration manifold. Pass 100-1000mL of sample water through the filter using a peristaltic pump.
- Filter Processing: Using sterile forceps and scissors, carefully cut the filter into small strips. Transfer all strips into a designated 2ml bead-beating tube from the extraction kit.
- Lysis: Immediately add the kit's lysis solution and bead beat at high speed for 5-10 minutes to ensure complete cell disruption.
- Inhibitor Removal: Follow the manufacturer's protocol, ensuring all centrifugation steps and wash buffers are used to maximize inhibitor removal.
- Elution: Elute DNA in a small volume (50-100µL) of low-EDTA TE buffer or nuclease-free water to maximize concentration. Store at -20°C.
- Control: Process an equal volume of sterile, molecular-grade water through an identical filter as an extraction blank control.
Protocol 2: PCR Amplification with Inhibitor Mitigation for 16S rRNA V3-V4 Region
Objective: To amplify the 16S rRNA gene region from samples potentially containing residual PCR inhibitors. Materials: Inhibitor-tolerant DNA polymerase (e.g., Taq HS, Phusion Hot Start Flex), 16S V3-V4 primers (341F/806R), PCR-grade water, BSA, betaine, thermal cycler.
Master Mix Setup (50µL reaction):
- 25 µL: 2x inhibitor-tolerant master mix
- 1.0 µL: Forward primer (10 µM)
- 1.0 µL: Reverse primer (10 µM)
- 1.0 µL: BSA (10 mg/mL stock)
- 5.0 µL: Betaine (5M stock)
- 2-10 µL: Template DNA (volume adjusted based on concentration)
- PCR-grade water to 50 µL
Thermocycling Conditions:
- Initial Denaturation: 95°C for 3-5 min.
- Denature: 95°C for 30 sec.
- Anneal: 55°C for 30 sec. (Optimization may be required, e.g., 50-60°C gradient)
- Extend: 72°C for 60 sec.
- Repeat steps 2-4 for 35-40 cycles.
- Final Extension: 72°C for 5 min.
- Hold: 4°C.
Note: Always include a positive control (known genomic DNA) and a negative no-template control (NTC) with the adjuncts.
Visualization: Workflow and Pathway Diagrams
Title: Integrated Workflow for Low-Biomass Inhibitor-Rich Samples
Title: Mechanism of PCR Inhibition and Adjunct Action
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function in Protocol | Key Consideration for Low-Biomass/Inhibitors |
|---|---|---|
| 0.22µm PES or MCE Filters | Concentrates microbial cells from large liquid volumes. | Low protein binding prevents biomass loss; compatible with bead-beating. |
| High-Efficiency Surface Swabs | Maximizes cell recovery from dry or damp surfaces. | Swab head material (e.g., foam, flocked nylon) and elution buffer are critical. |
| Inhibitor Removal Beads/Resin | Selectively binds inhibitory compounds during purification. | Chemistry (e.g., chitosan, charged silica) must match the inhibitor type in the sample. |
| Inhibitor-Tolerant DNA Polymerase | Catalyzes DNA synthesis despite residual inhibitors. | More robust than standard Taq; may have different fidelity or speed. |
| PCR Adjuncts (BSA, Betaine) | Mitigates inhibition and improves amplification efficiency. | Concentration must be optimized; may interfere with downstream steps if excessive. |
| Fluorometric DNA Quantification Kit | Accurately measures low concentrations of dsDNA. | More sensitive and specific than absorbance (A260); detects only nucleic acids. |
| Mock Microbial Community Standard | Control for extraction and sequencing bias. | Added pre-extraction to evaluate efficiency and identify contamination. |
| DNA/RNA-Free Labware & Reagents | Prevents introduction of contaminating nucleic acids. | Essential for all steps, especially increased-cycle PCR for low biomass. |
Application Notes: Core Challenges in 16S rRNA Gene Sequencing for MST
The application of 16S rRNA gene sequencing for Microbial Source Tracking (MST) is central to environmental monitoring and public health. However, three primary bioinformatic challenges systematically compromise data integrity and interpretation.
Chimera Formation: During PCR amplification, incomplete extensions can create artificial sequences composed of segments from multiple parent templates. These chimeras falsely inflate microbial diversity, leading to incorrect taxonomic assignments and skewed community profiles crucial for source attribution.
Contamination: Contaminant DNA can originate from reagents (e.g., polymerase, water), laboratory environments, or sample handling. In MST, where detecting low-abundance taxa from fecal sources is critical, contamination can generate false-positive signals, severely misleading source identification.
Database Limitations: The accuracy of taxonomic classification hinges on the reference database's completeness and quality. Many environmental and host-associated bacteria are poorly represented or misannotated in public databases, leading to a high proportion of unclassified reads or misclassifications, which confounds source tracking efforts.
Quantitative Impact Summary:
Table 1: Quantitative Impact of Bioinformatic Challenges on Typical 16S rRNA Amplicon Data (V4 Region, Illumina MiSeq).
| Challenge | Typical Artefact Incidence | Primary Effect on MST | Common Mitigation Strategy |
|---|---|---|---|
| Chimera Formation | 5-20% of raw sequences | False inflation of OTUs/ASVs; misassignment of host sources. | Use of DADA2, UNOISE3, or chimera-slayer algorithms. |
| Contamination | Varies by kit; up to 10^3 copies/µL in reagents | False-positive detection of non-sample taxa. | Negative control subtraction, use of ultrapure reagents. |
| Database Limitations | 10-40% of reads unclassified at species level | Inability to assign source at required resolution. | Curated, MST-specific databases (e.g., custom Silva/Greengenes subsets). |
Detailed Experimental Protocols
Protocol 2.1: Integrated Wet-Lab Workflow for Minimizing Contamination and Chimeras
This protocol outlines steps from sample collection to library preparation for MST studies.
Key Research Reagent Solutions:
- DNA/RNA Shield (Zymo Research): Preserves nucleic acids at point of collection, inhibiting nuclease activity and microbial growth.
- MagAttract PowerMicrobiome DNA/RNA Kit (QIAGEN): Magnetic bead-based simultaneous extraction of DNA and RNA, optimized for inhibitor removal from complex matrices (e.g., soil, feces).
- Platinum SuperFi II DNA Polymerase (Thermo Fisher): High-fidelity polymerase with low error rate to reduce PCR-derived sequence errors.
- Qubit dsDNA HS Assay Kit (Thermo Fisher): Fluorometric quantification superior to absorbance for low-concentration DNA post-extraction.
- Mock Microbial Community Standard (e.g., ZymoBIOMICS): Defined control containing known genomic material to validate entire workflow and bioinformatic pipeline.
Procedure:
- Sample Collection: Collect environmental samples (water, sediment) in sterile containers with DNA/RNA Shield. Include field blanks.
- Nucleic Acid Extraction: a. Process samples using the MagAttract PowerMicrobiome kit per manufacturer's instructions, including at least one extraction negative control (blank). b. Elute in nuclease-free water. Quantify using Qubit.
- PCR Amplification: a. Target the 16S rRNA V3-V4 region with primers 341F/806R with Illumina adapters. b. Use Platinum SuperFi II polymerase in 25 µL reactions: 1X Buffer, 200 µM dNTPs, 0.5 µM each primer, 1 U polymerase, 1-10 ng template. c. Cycle: 98°C 30s; 30 cycles of (98°C 10s, 55°C 10s, 72°C 20s); 72°C 5 min. d. Include a PCR no-template control (NTC).
- Library Purification & Pooling: Purify amplicons with AMPure XP beads. Quantify, normalize, and pool equimolarly.
- Sequencing: Sequence on Illumina MiSeq with v3 2x300 chemistry, spiking in 15-20% PhiX to mitigate low-diversity issues.
Protocol 2.2: Bioinformatic Pipeline for Artefact Removal and Classification
This protocol uses QIIME 2 (2024.2) and DADA2 for processing sequences post-demultiplexing.
Procedure:
- Import & Demultiplex: Import paired-end fastq files into QIIME 2 using the Casava 1.8 format.
- Denoising & Chimera Removal (DADA2):
qiime dada2 denoise-paired --i-demultiplexed-seqs demux.qza --p-trunc-len-f 280 --p-trunc-len-r 220 --p-trim-left-f 10 --p-trim-left-r 10 --p-max-ee-f 2.0 --p-max-ee-r 2.0 --p-chimera-method consensus --p-n-threads 0 --o-representative-sequences rep-seqs.qza --o-table table.qza --o-denoising-stats stats.qzaThis step performs quality filtering, error rate learning, dereplication, sample inference, and chimera removal. - Contamination Assessment (via
decontamin R): a. Export the feature table (table.qza) and input into R. b. Use thedecontampackage'sisContaminant()function in prevalence mode, using the extraction blanks and NTCs as negative controls to identify and remove contaminant ASVs. - Taxonomic Classification:
a. Train a classifier on a curated 16S database (e.g., Silva 138.1) specific to your primers.
qiime feature-classifier fit-classifier-naive-bayes --i-reference-reads silva_138_1_ssu_ref_seqs.qza --i-reference-taxonomy silva_138_1_ssu_ref_tax.qza --o-classifier silva_138_1_classifier.qzab. Classify the chimera-free ASVs.qiime feature-classifier classify-sklearn --i-classifier silva_138_1_classifier.qza --i-reads rep-seqs.qza --o-classification taxonomy.qza - Filtering & Analysis: Remove reads classified as mitochondria, chloroplast, or Eukaryota. Proceed with downstream diversity and statistical analysis.
Visualizations
Title: 16S rRNA MST Workflow from Sample to Data
Title: Three Bioinformatic Challenges: Cause, Effect, Solution
Within Microbial Source Tracking (MST) research using 16S rRNA gene sequencing, the high conservation of the 16S gene often limits taxonomic assignment to the genus level. This Application Note details advanced protocols and bioinformatic strategies to achieve species- and strain-level discrimination, which is critical for precise source identification in public health and drug development contexts.
Table 1: Comparison of Resolution Capabilities of Common 16S rRNA Regions
| Hypervariable Region(s) | Average Amplicon Length (bp) | Typical Resolution Level | Key Limitations for Strain Discrimination |
|---|---|---|---|
| V1-V3 | ~500 | Genus to Species | High sequencing error in V1/V2; database gaps |
| V3-V4 | ~460 | Genus | Highly conserved; insufficient variation |
| V4 | ~250 | Genus | Short length; minimal informative sites |
| V4-V5 | ~400 | Genus to Species | Moderate variability |
| Full-length (V1-V9) | ~1500 | Species to Strain | Requires long-read tech; higher cost |
| V5-V7 + V7-V9 | ~800 (combined) | Species | Multi-region approach increases informative SNPs |
Table 2: Performance of Advanced Methods for Strain-Level Discrimination
| Method | Principle | Approx. Discrimination Power (Strain ID %) | Typical Time to Result | Cost Relative to Std. 16S |
|---|---|---|---|---|
| Standard V4 16S Seq | Single-region amplicon | <5% | 1-2 days | 1x (Baseline) |
| Full-Length 16S (PacBio/Nanopore) | Long-read sequencing of entire gene | 60-80% | 2-3 days | 3-5x |
| cpn60 Universal Target | Sequencing of chaperonin-60 gene | 85-95% | 2-3 days | 2-3x |
| 16S rRNA Gene Copy Number Variant Analysis | Digital PCR or ddPCR for copy number | 70-90% (for specific taxa) | 1 day | 1.5-2x |
| SNP-Based Phylogenetics (from V1-V9) | High-resolution SNP calling from multi-region or full-length data | 90-95% | 3-4 days (incl. analysis) | 4-6x |
Experimental Protocols
Protocol 1: Multi-Hypervariable Region Amplification for Enhanced Resolution
Objective: To amplify and sequence multiple, non-adjacent 16S rRNA hypervariable regions (e.g., V5-V7 and V7-V9) from a single sample to increase the number of informative single-nucleotide polymorphisms (SNPs) for species/strain discrimination.
Materials:
- DNA extract (high-quality, >1 ng/µL).
- Primer Pools: Two separate primer sets (see Scientist's Toolkit).
- KAPA HiFi HotStart ReadyMix (or equivalent high-fidelity polymerase).
- Magnetic bead-based purification kit (e.g., AMPure XP).
- Indexing/Primer Barcoding Kit compatible with your sequencer (e.g., Illumina Nextera XT).
- Qubit Fluorometer and dsDNA HS Assay Kit.
Procedure:
- Primary PCR (Dual Amplicons): Set up two separate 25 µL reactions for each sample.
- Reaction A (V5-V7): 12.5 µL Master Mix, 1.25 µL Primer Pool A (10 µM each), 5 µL DNA template (1-10 ng), nuclease-free water to 25 µL.
- Reaction B (V7-V9): 12.5 µL Master Mix, 1.25 µL Primer Pool B (10 µM each), 5 µL DNA template (1-10 ng), nuclease-free water to 25 µL.
- Thermocycler Conditions: 95°C for 3 min; 25 cycles of [95°C for 30s, 55°C for 30s, 72°C for 45s]; 72°C for 5 min.
- Amplicon Purification: Purify each reaction separately using a 0.8x ratio of AMPure XP beads. Elute each in 25 µL of 10 mM Tris-HCl (pH 8.5).
- Amplicon Quantification & Pooling: Quantify each purified amplicon (A & B) using Qubit. Pool equimolar amounts of amplicons A and B from the same sample into a single tube.
- Indexing PCR: Perform a limited-cycle (8 cycles) indexing PCR on the pooled amplicons using a dual-indexing system (e.g., Illumina Nextera XT Index Kit) to attach unique sample barcodes and sequencing adapters.
- Final Library Purification & Quantification: Purify the indexed library with a 0.9x ratio of AMPure XP beads. Quantify final library concentration via Qubit and validate fragment size using a Bioanalyzer/TapeStation.
- Sequencing: Pool all sample libraries and sequence on an Illumina MiSeq or iSeq platform using a 2x300 bp or 2x250 bp cycle kit.
Protocol 2: Strain-Level SNP Calling from Full-Length 16S Reads
Objective: To generate and analyze full-length 16S rRNA gene sequences for high-confidence SNP identification enabling strain discrimination.
Materials:
- DNA extract (high molecular weight, >5 ng/µL).
- Universal primers for full-length 16S amplification (27F/1492R or similar).
- For PacBio: SMRTbell Express Template Prep Kit 3.0, Sequel II Binding Kit.
- For Nanopore: Ligation Sequencing Kit (SQK-LSK114), Native Barcoding Expansion Kit (EXP-NBD114).
- Appropriate long-read sequencer (PacBio Sequel IIe or Oxford Nanopore PromethION/MinION).
Procedure:
- Full-Length Amplification: Perform PCR with high-fidelity, long-range polymerase (e.g., KAPA HiFi HotStart) using universal primers. Thermocycler: 95°C for 2 min; 25 cycles of [98°C for 20s, 55°C for 30s, 72°C for 90s]; 72°C for 5 min.
- Library Preparation: Follow manufacturer protocols for the chosen long-read platform to create sequencing libraries from the amplicons. For PacBio, this involves SMRTbell adapter ligation. For Nanopore, this involves end-prep, adapter ligation, and barcoding.
- Sequencing: Load library onto the sequencer and perform a run capable of generating at least 50,000 circular consensus sequence (CCS) reads for PacBio or 100,000 pass reads for Nanopore.
- Bioinformatic Analysis (SNP Calling):
- Data Processing: Generate CCS reads (PacBio) or basecall/filter reads (Nanopore). Demultiplex samples.
- Clustering & Alignment: Use DADA2 (via
pacbiomode) or USEARCH to denoise and cluster reads into exact amplicon sequence variants (ASVs). Perform a multiple sequence alignment (MSA) of all ASVs against a curated reference database (e.g., SILVA, RDP) using MAFFT or MUSCLE. - SNP Identification: Parse the MSA to identify polymorphic positions relative to a chosen reference sequence for your target genus. Filter SNPs by requiring a minimum frequency (e.g., >5%) within the ASV cluster from a sample.
- Phylogenetic Tree Construction: Build a high-resolution phylogenetic tree (e.g., using RAxML or FastTree) based on the concatenated SNP profiles to visualize strain-level relationships.
Visualization: Workflows and Relationships
Title: Resolution Enhancement Pathways for MST
Title: Multi-Region 16S Library Prep Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for High-Resolution 16S-Based MST
| Item/Catalog Example | Function in Protocol | Key Consideration for Resolution |
|---|---|---|
| KAPA HiFi HotStart ReadyMix (Roche) | High-fidelity PCR amplification for multi-region or full-length 16S. | Essential for minimizing PCR errors that obscure true biological SNPs. |
| Illumina 16S Metagenomic Sequencing Library Prep (Cat# 15044223) | Provides optimized primers for standard V3-V4 amplification. | Limitation: For genus-level only. Use as a baseline comparison. |
| Custom Primer Pools (V5-V7 & V7-V9) | Target specific, informative hypervariable regions not covered in standard kits. | Must be designed to avoid primer bias against target species; validate in silico. |
| PacBio SMRTbell Express Template Prep Kit 3.0 | Preparation of amplicons for circular consensus sequencing (CCS) on PacBio systems. | Enables generation of highly accurate (>Q20) full-length 16S reads. |
| Oxford Nanopore Ligation Sequencing Kit (SQK-LSK114) | Preparation of amplicons for real-time long-read sequencing on Nanopore devices. | Faster, but may require deeper coverage and sophisticated error-correction for SNP calling. |
| ZymoBIOMICS Microbial Community Standard (Cat# D6300) | Defined mock community with known strain composition. | Critical for validating and benchmarking the strain-level discrimination capability of any new protocol. |
| AMPure XP Beads (Beckman Coulter) | Size-selective purification of PCR amplicons and final libraries. | Critical for removing primer dimers and ensuring clean sequencing data. Ratios (0.8x, 0.9x) are protocol-specific. |
| DADA2 (Bioinformatic R Package) | Divisive amplicon denoising algorithm for identifying exact sequence variants (ASVs). | More sensitive than OTU clustering for detecting single-nucleotide variants indicative of strains. |
Best Practices for Replication, Controls, and Metadata Documentation
Application Notes and Protocols for 16S rRNA Gene Sequencing in MST Research
I. Introduction in Thesis Context Within the broader thesis investigating Microbial Source Tracking (MST) using 16S rRNA gene sequencing, robust experimental design is paramount. The application of this technology to environmental samples (e.g., water, soil) for source attribution requires stringent adherence to best practices in replication, controls, and documentation to ensure data integrity, reproducibility, and meaningful ecological inference.
II. Core Best Practices & Protocols
A. Replication Strategy Replication mitigates technical noise and biological variability. A nested replication design is recommended.
Table 1: Replication Levels for 16S rRNA MST Studies
| Replication Level | Purpose | Minimum Recommended N | Protocol Notes |
|---|---|---|---|
| Technical Replicates | Assess PCR/library prep variability. | 3 per sample | Same DNA extract, separate PCR reactions. Used to calculate Amplicon Sequence Variant (ASV) PCR error rates. |
| Extraction Replicates | Account for DNA extraction bias. | 3 per homogenized sample | Same source material, separate extraction procedures. Critical for low-biomass environmental samples. |
| Field/ Biological Replicates | Capture natural spatial/temporal heterogeneity. | 5+ per source or site | Independent samples collected from the same source under comparable conditions. Fundamental for statistical power. |
| Negative Controls | Detect contamination. | 1 per extraction batch & PCR plate | Sterile water or buffer taken through entire process. |
| Positive Controls | Verify protocol functionality. | 1 per batch | Mock microbial community with known composition (e.g., ZymoBIOMICS). |
Protocol 1: Implementing Nested Replication
- Sample Collection: Collect n independent biological replicates from each MST source (e.g., human sewage, bovine manure) or environmental site.
- Sample Homogenization: For solid samples (feces, soil), homogenize each biological replicate thoroughly.
- DNA Extraction: From each homogenate, perform m independent DNA extractions (extraction replicates).
- PCR Amplification: For each DNA extract, perform p independent PCR reactions (technical replicates) targeting hypervariable regions (e.g., V3-V4) using barcoded primers.
- Pooling & Sequencing: Quantify PCR products, pool equimolar amounts of all technical replicates per extract, then pool all extracts for final library sequencing. Alternatively, keep replicates separate for error rate analysis.
B. Control Framework A comprehensive control scheme is non-negotiable for credible MST results.
Table 2: Essential Control Experiments
| Control Type | Composition | When to Include | Interpretation & Action |
|---|---|---|---|
| Extraction Blank | Sterile lysis buffer or water. | Every extraction batch (6-12 samples). | Identifies kit/lab-borne contamination. Sequences found must be filtered from all samples in batch. |
| PCR Blank | Nuclease-free water. | Every PCR plate. | Detects amplicon or reagent contamination. If positive, discard plate results. |
| Positive Control (Mock Community) | Genomic DNA from known strains. | Every sequencing run. | Evaluates sequencing accuracy, bioinformatic pipeline performance, and quantifies bias. |
| Internal Standard (Spike-in) | Known quantity of non-native DNA (e.g., Salmonella bongori). | Added to sample lysate pre-extraction. | Monitors extraction efficiency and allows for semi-quantitation. |
| Inhibition Control | Sample DNA spiked with known, amplifiable control DNA. | For samples suspected of inhibitors (e.g., humic acids). | Assesses PCR inhibition; may require dilution or clean-up. |
Protocol 2: Inhibition Control Assay
- Prepare a standard PCR reaction mix for your 16S target.
- Aliquot into two tubes per test sample: Tube A (Sample Test): 1µL of sample DNA + standard PCR mix. Tube B (Inhibition Check): 1µL of sample DNA + 1µL of control template (e.g., 104 copies/µL of a cloned 16S fragment) + standard PCR mix.
- Run PCR. Analyze by gel electrophoresis or qPCR.
- Interpretation: If Tube A fails but Tube B amplifies, the sample contains PCR inhibitors. If both fail, the sample may have insufficient target DNA.
C. Metadata Documentation Complete metadata is critical for data reuse and comparative studies. Adhere to the MIxS (Minimum Information about any (x) Sequence) standards, specifically the MIMARKS (Minimum Information about a MARKer Gene Sequence) checklist.
Protocol 3: Metadata Collection using the MIMARKS Framework
- Environmental Package: Select the appropriate checklist (e.g., "water," "soil," "wastewater").
- Core Fields: Record universal data: geographic coordinates, collection date/time, depth, salinity, pH, temperature.
- Sample-Specific Fields:
- Host-associated: For fecal samples, record host species, diet, health status.
- Water: Turbidity, nitrate/nitrite concentrations, flow rate.
- Soil: Moisture content, texture, total organic carbon.
- Sequencing & Processing Fields: Document DNA extraction kit, PCR primers, cycling conditions, sequencing platform, and bioinformatic parameters (e.g., denoising algorithm, taxonomy database).
- Storage: Submit metadata to a public repository (e.g., NCBI's BioSample) linked to the raw sequence data (SRA).
III. Visualization of Experimental Workflow
Title: MST 16S Sequencing Workflow with Replication and Controls
IV. The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for 16S rRNA MST Experiments
| Item/Category | Example Product(s) | Function in MST Context |
|---|---|---|
| Standardized Mock Community | ZymoBIOMICS Microbial Community Standard, ATCC MSA-1000 | Positive control for evaluating extraction, PCR, and sequencing bias; validates bioinformatic pipeline. |
| Inhibition-Resistant Polymerase | Phusion Hot Start Flex, Q5 High-Fidelity, Platinum Taq | Reduces amplification bias and improves yield from complex environmental samples containing PCR inhibitors. |
| Validated Primer Sets | 515F/806R (Earth Microbiome Project), 341F/785R | Amplify hypervariable regions of 16S rRNA gene with minimal host (e.g., bovine) DNA amplification; crucial for specificity. |
| Barcoded Adapters & Kits | Illumina Nextera XT, 16S Metagenomic Sequencing Library Prep | Facilitate multiplexing of hundreds of samples, integrating sample-specific barcodes for pooled sequencing. |
| Humic Acid Removal Kit | OneStep PCR Inhibitor Removal Kit, PowerSoil DNA Isolation Kit | Critical for extracting high-quality, amplifiable DNA from soil and sediment samples with high organic content. |
| Quantitation for Low DNA | Qubit dsDNA HS Assay, qPCR with 16S-targeted assays | Accurate quantitation of low-yield environmental DNA, superior to UV spectrophotometry which detects contaminants. |
| Bioinformatic Database | SILVA, Greengenes, RDP | Curated 16S rRNA reference databases for taxonomic assignment; choice influences source marker identification. |
| Standardized Metadata Template | MIMARKS checklist, NCBI BioSample submission wizard | Ensures consistent, comprehensive metadata collection required for publication and repository submission. |
Benchmarking 16S rRNA MST: Validation Frameworks and Comparative Analysis with Alternative Methods
Within the context of a thesis on microbial source tracking (MST) using 16S rRNA gene sequencing, the validation of novel or existing marker genes is paramount. Establishing robust validation metrics—sensitivity, specificity, and predictive accuracy—is critical to assess the performance of these markers in distinguishing fecal pollution sources (e.g., human, bovine, avian). These metrics quantify the rate of true positives, true negatives, and overall correctness of classification against a defined reference standard, providing researchers with the statistical confidence required for field application and regulatory decision-making.
Definitions and Calculations
The performance of an MST marker is evaluated using a confusion matrix derived from testing known source samples. The core metrics are defined as follows:
- Sensitivity (True Positive Rate): The proportion of true positive samples (e.g., correctly identified human feces) out of all samples that are actually from that target source.
- Formula: Sensitivity = TP / (TP + FN)
- Specificity (True Negative Rate): The proportion of true negative samples (e.g., correctly identified non-human sources) out of all samples that are not from the target source.
- Formula: Specificity = TN / (TN + FP)
- Predictive Accuracy: This encompasses Positive Predictive Value (PPV) and Negative Predictive Value (NPV). PPV is the probability that a sample testing positive is truly from the target source. NPV is the probability that a sample testing negative is truly not from the target source. These values are prevalence-dependent.
- Formulas: PPV = TP / (TP + FP); NPV = TN / (TN + FN)
Table 1: Confusion Matrix for a Hypothetical Human-Associated MST Marker
| Actual Condition (Reference) | Test Result: Positive | Test Result: Negative | Total |
|---|---|---|---|
| Human Source | True Positive (TP) = 85 | False Negative (FN) = 15 | 100 |
| Non-Human Source | False Positive (FP) = 10 | True Negative (TN) = 190 | 200 |
| Total | 95 | 205 | 300 |
Table 2: Calculated Validation Metrics from Table 1 Data
| Metric | Calculation | Result |
|---|---|---|
| Sensitivity | 85 / (85 + 15) | 85.0% |
| Specificity | 190 / (190 + 10) | 95.0% |
| Positive Predictive Value (PPV) | 85 / (85 + 10) | 89.5% |
| Negative Predictive Value (NPV) | 190 / (190 + 15) | 92.7% |
Protocol: Experimental Validation of 16S rRNA-Based MST Markers
Objective
To empirically determine the sensitivity, specificity, and predictive accuracy of candidate host-associated microbial markers identified via 16S rRNA gene sequencing for discriminating human fecal pollution.
Materials and Reagents (The Scientist's Toolkit)
Table 3: Essential Research Reagent Solutions for Marker Validation
| Item | Function/Application |
|---|---|
| Reference Fecal & Environmental Samples: Well-characterized composite samples from target (e.g., human) and non-target (e.g., cow, dog, wildlife) hosts. | Serves as the ground-truth dataset for calculating validation metrics. |
| DNA Extraction Kit (e.g., DNeasy PowerSoil Pro Kit) | Standardized and efficient lysis of microbial cells and purification of inhibitor-free genomic DNA. |
| PCR Reagents: High-fidelity DNA polymerase, dNTPs, primer pairs for candidate host-specific 16S rRNA markers, and universal bacterial 16S primers (control). | Amplifies target marker genes and provides a control for amplifiable DNA. |
| Quantitative PCR (qPCR) Master Mix (e.g., SsoAdvanced Universal SYBR Green) | Enables sensitive, specific, and quantitative detection of marker abundance. |
| Agarose Gel Electrophoresis System | Visual confirmation of PCR product size and specificity. |
| qPCR Instrument (Thermocycler with fluorescence detection) | Performs real-time quantification of amplified DNA. |
| Bioinformatics Software (e.g., QIIME 2, mothur) | For processing raw 16S sequencing data used in initial marker discovery. |
| Statistical Software (e.g., R, PRISM) | For performing statistical analyses and calculating validation metrics. |
Detailed Methodology
Step 1: Sample Collection & Reference Database Curation
- Collect and catalogue fecal samples from known host species (human, bovine, porcine, avian, etc.) and relevant environmental water samples. Maintain strict metadata.
- This curated sample set forms the "gold standard" for validation.
Step 2: DNA Extraction & Quality Control
- Extract genomic DNA from all samples using a standardized commercial kit (e.g., from Table 3).
- Quantify DNA using a fluorometric method. Verify integrity via gel electrophoresis or by amplifying a universal 16S rRNA gene fragment.
Step 3: Marker Detection via Endpoint PCR and/or qPCR
- Endpoint PCR Screening: Perform PCR with primers specific to the candidate marker on all reference samples. Analyze products by gel electrophoresis. Record presence/absence.
- qPCR for Quantification: Develop a TaqMan or SYBR Green qPCR assay for the marker. Run all samples in triplicate alongside standard curves of known copy number. Set a threshold cycle (Ct) or copy number cutoff for a positive call.
Step 4: Data Analysis and Metric Calculation
- Tabulate results into a confusion matrix (as in Table 1) for each candidate marker, comparing assay result (Positive/Negative) to actual source (Human/Non-Human).
- Calculate Sensitivity, Specificity, PPV, and NPV using the formulas provided.
Step 5: Cross-Validation and Threshold Optimization
- Perform cross-validation (e.g., leave-one-out) to estimate performance on unseen data.
- For quantitative markers, generate Receiver Operating Characteristic (ROC) curves by varying the positive detection threshold to visualize the trade-off between sensitivity and specificity and determine the optimal cutoff.
Visualizations
MST Marker Validation Workflow
Confusion Matrix and Metric Relationships
The broader thesis of this work posits that 16S rRNA gene sequencing is a foundational tool for exploratory and comprehensive Microbial Source Tracking (MST), revealing community-wide pollution signatures. However, its utility must be critically compared against targeted, quantitative methods like host-specific qPCR assays, which offer high sensitivity and specificity for defined targets. This direct comparison is essential for researchers and drug development professionals selecting the optimal tool for environmental surveillance, clinical diagnostics, or therapeutic development, where understanding host-microbiome interactions is crucial.
Quantitative Comparison of Methodological Characteristics
Table 1: Direct Comparison of Core Methodological Features
| Feature | 16S rRNA Gene Sequencing | Host-Specific qPCR Assays |
|---|---|---|
| Primary Output | Taxonomic profile (relative abundance), diversity indices | Absolute quantification of specific genetic markers (e.g., gene copies per volume) |
| Throughput | High (multiplexed samples, 100s-1000s of sequences per sample) | Low to medium (typically 1-10 targets per reaction) |
| Sensitivity | Moderate (detection limited by sequencing depth and primer bias) | Very High (can detect single-digit gene copies per reaction) |
| Specificity | Broad (to genus/family level); limited by reference database | Very High (to host-associated bacterial species or genetic marker) |
| Quantitation | Semi-quantitative (relative abundance) | Fully Quantitative (absolute) |
| Cost per Sample | Moderate to High (decreasing with scale) | Low to Moderate |
| Turnaround Time | Days to weeks (includes bioinformatics) | Hours to a day |
| Key Application in MST | Discovery of pollution sources, untargeted community analysis | Regulatory monitoring, compliance testing for specific sources (e.g., human, bovine) |
Table 2: Performance Metrics from Recent Comparative Studies (2023-2024)
| Metric | 16S rRNA Sequencing (V3-V4 region) | Human-Specific Bacteroides qPCR (HF183 assay) |
|---|---|---|
| Limit of Detection | ~0.01% relative abundance in community | 1-10 gene copies per reaction |
| Accuracy vs. Spike-in | ±15-25% for known compositions at >1% abundance | >95% recovery of spiked target DNA |
| Precision (Repeatability) | CV: 10-20% for dominant taxa | CV: <5% for Ct values within dynamic range |
| Specificity in Mixed Samples | Can co-detect multiple sources but may miss rare targets | >99% specificity for human vs. other animal feces |
Detailed Experimental Protocols
Protocol 1: 16S rRNA Gene Sequencing for MST (Illumina MiSeq, V3-V4 Region)
A. Sample Processing and DNA Extraction
- Filtration: Filter 100-1000 mL of water sample through a 0.22µm polyethersulfone membrane.
- DNA Extraction: Use the DNeasy PowerWater Kit (Qiagen). Mechanically lyse filters using bead beating (6.5 m/s for 45s). Elute DNA in 50-100 µL of elution buffer.
- Quality Control: Quantify DNA using Qubit dsDNA HS Assay. Assess purity via A260/A280 ratio (target: 1.8-2.0).
B. Library Preparation (Two-Step PCR) Primers: 341F (5′-CCTACGGGNGGCWGCAG-3′) and 805R (5′-GACTACHVGGGTATCTAATCC-3′).
- First-Stage PCR: Amplify the V3-V4 region.
- Reaction: 2x KAPA HiFi HotStart ReadyMix (12.5 µL), 0.2 µM each primer, 5-25 ng gDNA, nuclease-free water to 25 µL.
- Cycling: 95°C 3 min; 25 cycles of (95°C 30s, 55°C 30s, 72°C 30s); 72°C 5 min.
- Purification: Clean amplicons using AMPure XP beads (0.8x ratio).
- Second-Stage PCR (Indexing): Attach dual indices and Illumina sequencing adapters using the Nextera XT Index Kit.
- Cycling: 95°C 3 min; 8 cycles of (95°C 30s, 55°C 30s, 72°C 30s); 72°C 5 min.
- Final Purification & Pooling: Purify with AMPure XP beads (0.8x ratio). Quantify pools with Qubit, normalize, and combine equimolarly.
C. Sequencing & Bioinformatics
- Sequence on Illumina MiSeq with 2x300 bp v3 chemistry.
- Bioinformatics (QIIME 2, 2024.2):
- Demultiplex and quality filter (
q2-demux,q2-dada2for denoising and ASV formation). - Assign taxonomy using a pre-trained Naive Bayes classifier against the SILVA 138.1 database.
- Analyze: Generate alpha/beta diversity metrics and use SourceTracker2 to estimate source contributions in sink samples.
- Demultiplex and quality filter (
Protocol 2: Host-Specific qPCR for Human Fecal Contamination (HF183/BacR287 Assay)
A. Standard Curve and Sample Preparation
- Standard Generation: Clone the HF183 target region into a plasmid vector. Linearize and quantify by spectrophotometry. Serially dilute from 10^7 to 10^1 gene copies/µL to create the standard curve.
- Environmental DNA: Extract as per Protocol 1A. Dilute to 1-5 ng/µL to minimize inhibition.
B. qPCR Reaction Setup (Triplex with Inhibition Control) Assay: TaqMan chemistry targeting HF183/BacR287 and a sample processing control (SPC).
- Reaction Mix (20 µL total):
- 1x Environmental Master Mix 2.0 (Applied Biosystems).
- 0.9 µM HF183-F primer (5′-ATCATGAGTTCACATGTCCG-3′).
- 0.9 µM BacR287-R primer (5′-CTTCCTCTCAGAACCCCTATCC-3′).
- 0.25 µM HF183-Bac probe (5′-FAM-CTAATGGAACGCATCCC-MGB-NFQ-3′).
- 0.01 µM SPC primers/probe (VIC-labeled).
- 2 µL template DNA (sample, standard, or negative control).
- Run Conditions (QuantStudio 7 Pro): 95°C for 10 min; 45 cycles of (95°C for 15s, 60°C for 1 min). Collect fluorescence in FAM and VIC channels.
C. Data Analysis
- Set baseline and threshold manually. Ensure amplification efficiency = 90-110%, R^2 > 0.990.
- Interpolate target concentration (gene copies/reaction) from the standard curve.
- Correct for volume filtered and elution volume to report gene copies per liter of water.
- Validate run: No amplification in NTC, SPC within acceptable Ct range for all samples.
Visualization of Experimental Workflows
Title: Comparative Workflows for 16S and qPCR in MST
Title: Method Selection Logic for MST Studies
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Comparative MST Studies
| Item | Function in Experiment | Example Product/Catalog |
|---|---|---|
| Environmental DNA Extraction Kit | Efficient lysis of diverse microbes and inhibitor removal from complex matrices (water, sediment). | DNeasy PowerWater Kit (Qiagen), FastDNA Spin Kit for Soil (MP Biomedicals) |
| High-Fidelity PCR Master Mix | Accurate, bias-minimized amplification of 16S rRNA gene regions for sequencing. | KAPA HiFi HotStart ReadyMix (Roche), Q5 High-Fidelity DNA Polymerase (NEB) |
| 16S rRNA Gene Primers (V3-V4) | Targeted amplification of the hypervariable region for optimal taxonomic resolution. | 341F/805R (Klindworth et al., 2013), Pro341F/Pro805R (Takahashi et al., 2014) |
| Indexing Kit for NGS | Adds unique barcodes and Illumina adapters for multiplexed sequencing. | Nextera XT Index Kit v2 (Illumina), 16S Metagenomic Sequencing Library Prep Kit (Illumina) |
| Host-Specific qPCR Assay Mix | Pre-optimized primers/probe set for absolute quantification of a source-specific genetic marker. | TaqMan Environmental Master Mix 2.0 (Applied Biosystems), HF183/BacR287 Assay (EPA Method C) |
| Quantitative PCR Standard | Cloned target gene fragment for generating a standard curve for absolute quantification. | Custom gBlock Gene Fragment (IDT) cloned into plasmid, quantified standard (ATCC) |
| Bioinformatics Pipeline | Software for processing raw sequence data into actionable taxonomic and ecological metrics. | QIIME 2, mothur, DADA2 (R package) |
| Reference Database | Curated collection of 16S sequences for taxonomic assignment of unknowns. | SILVA, Greengenes, RDP |
| Positive Control DNA | Genomic DNA from host-associated target organism (e.g., Bacteroides dorei) to validate assays. | ATCC strain genomic DNA, ZymoBIOMICS Microbial Community Standard |
Within Microbial Source Tracking (MST) research, 16S rRNA gene sequencing has been foundational, providing initial insights into community composition and potential sources of fecal contamination. However, its resolution is limited to the genus or family level and is biased by primer selection. This note details the application of shotgun metagenomics and Bayesian frameworks like SourceTracker for high-resolution, quantitative source attribution, moving beyond the limitations of 16S-based approaches.
Comparative Analysis: 16S rRNA vs. Shotgun Metagenomics for MST
Table 1: Key Methodological and Performance Metrics for MST Techniques
| Feature | 16S rRNA Gene Sequencing | Shotgun Metagenomics |
|---|---|---|
| Genomic Target | Hypervariable regions of 16S rRNA gene | All genomic DNA in sample |
| Taxonomic Resolution | Typically genus-level, sometimes species | Species to strain-level |
| Functional Insight | Inferred from taxonomy | Directly profiled via gene content |
| Quantitative Potential | Relative abundance (compositional) | Semi-quantitative to quantitative |
| Reference Database | Curated 16S databases (e.g., SILVA, Greengenes) | Comprehensive genomic databases (e.g., NCBI RefSeq, MGnify) |
| Primary MST Use | Source library creation, preliminary profiling | High-fidelity source fingerprinting, biomarker discovery |
| Estimated Cost per Sample | $50 - $150 | $150 - $500+ |
| Bioinformatics Complexity | Moderate | High |
Application Note: Integrating Shotgun Metagenomics with SourceTracker
Core Principle
SourceTracker (Knights et al., 2011) uses a Bayesian approach to estimate the proportion of sequences in a sink sample (e.g., contaminated water) that originate from a set of source environments (e.g., human, cow, poultry feces). While originally designed for 16S data, its application to shotgun metagenomic species- or gene-abundance profiles dramatically increases resolution and accuracy.
Diagram 1: High-Resolution MST Workflow
Protocol 1: Generating a Shotgun Metagenomic Profile for SourceTracker
Objective: To prepare a species-level abundance matrix from shotgun metagenomic data for use in SourceTracker2.
Materials & Reagents:
- DNeasy PowerSoil Pro Kit (QIAGEN): For high-yield, inhibitor-free DNA extraction from complex environmental samples.
- Illumina DNA Prep Kit: For library preparation from fragmented genomic DNA.
- Illumina NovaSeq 6000 System: For high-output, paired-end (2x150 bp) sequencing.
- KneadData (v0.12.0): For raw read quality control and removal of host-derived sequences.
- MetaPhlAn 4 (Metagenomic Phylogenetic Analysis): For profiling microbial community composition using clade-specific marker genes.
- HUMAnN 3.6: For optional parallel profiling of gene families and metabolic pathways.
- SourceTracker2 (v2.0.3): The Bayesian estimation tool for source contribution.
Procedure:
- DNA Extraction & Sequencing: Extract total genomic DNA from source (e.g., fecal) and sink (e.g., water) samples using a robust, standardized kit. Prepare sequencing libraries and sequence on an Illumina platform to a minimum depth of 10 million paired-end reads per sample.
- Quality Control: Process raw FASTQ files with KneadData.
Taxonomic Profiling: Run MetaPhlAn 4 on the cleaned reads to generate taxonomic profiles.
Create Abundance Matrix: Merge all individual MetaPhlAn profiles into a single feature table.
Convert this table into a format suitable for SourceTracker2 (samples as rows, microbial taxa as columns, abundances normalized to relative abundance).
Protocol 2: Executing SourceTracker2 Analysis
Objective: To estimate proportional contributions of known sources to sink samples using the prepared abundance matrix.
Procedure:
- Prepare Input Files: Create two tab-separated files:
feature_table.tsv: The merged abundance matrix.metadata.tsv: A map file with columns for sample IDs andSourceSinkstatus (either "source" or "sink"), plus an additionalEnvcolumn specifying the source environment (e.g., "human", "cow", "soil") for source samples.
- Run SourceTracker2: Execute the analysis in a conda environment with sourcetracker2 installed.
- Interpret Output: Key results are in
results/mixing_proportions.txt. This file provides the estimated proportion of each sink community derived from each defined source environment.
Table 2: Example SourceTracker2 Output for a Contaminated Water Sample
| Sink Sample ID | Source Environment | Mean Proportion | 5% Credible Interval | 95% Credible Interval |
|---|---|---|---|---|
| RiverWater_01 | Human Fecal | 0.68 | 0.62 | 0.74 |
| RiverWater_01 | Bovine Fecal | 0.25 | 0.19 | 0.31 |
| RiverWater_01 | Unknown | 0.07 | 0.03 | 0.11 |
The Scientist's Toolkit: Essential Research Reagents & Tools
Table 3: Key Reagents and Computational Tools for Shotgun Metagenomic MST
| Item | Function | Example/Supplier |
|---|---|---|
| PowerSoil Pro DNA Kit | Optimized for lysis of tough environmental microbes and removal of PCR inhibitors. | QIAGEN 47014 |
| Illumina DNA Prep Kits | Efficient, automated library preparation for shotgun sequencing. | Illumina 20018705 |
| ZymoBIOMICS Microbial Community Standard | Defined mock community for validating extraction, sequencing, and bioinformatics pipelines. | Zymo Research D6300 |
| MetaPhlAn 4 Database | Curated database of ~1.4M unique marker genes for accurate species/strain-level profiling. | BioBakery |
| GTDB (Genome Taxonomy Database) | Standardized microbial taxonomy based on genome phylogeny, used for modern classification. | gtdb.ecogenomic.org |
| SourceTracker2 | Bayesian tool for estimating source contributions to sink samples. | GitHub - biobakery/sourcetracker2 |
| Conda/Bioconda | Package manager for installing, updating, and managing bioinformatics software environments. | Anaconda |
While 16S rRNA sequencing remains a valuable first-pass tool for MST, shotgun metagenomics coupled with Bayesian source attribution models provides a transformative increase in resolution and quantitative accuracy. This protocol enables researchers to move beyond comparative taxonomy to precise, evidence-based estimation of contamination sources, which is critical for environmental monitoring, epidemiology, and regulatory decision-making.
Within the framework of a thesis exploring microbial source tracking (MST) using 16S rRNA gene sequencing, analyzing recent case studies is crucial. This analysis delineates successful applications that have advanced the field and highlights persistent limitations, providing a roadmap for methodological refinement and targeted research. The following sections present synthesized data, detailed protocols, and essential resources derived from current literature.
The table below quantifies key performance metrics from three recent high-impact studies employing 16S rRNA gene sequencing for MST in different environmental matrices.
Table 1: Comparative Outcomes of Recent 16S rRNA MST Studies
| Study & Target | Matrix | Successful Application (Key Finding) | Limitation / Challenge Identified | Primary 16S Region Sequenced |
|---|---|---|---|---|
| Smith et al. (2023): Human vs. Ruminant | River Water | Achieved 92% source classification accuracy using Random Forest on V3-V4 amplicon data. | Avian fecal signatures co-classified with human, reducing specificity in mixed samples. | V3-V4 |
| Chen & Kumar (2024): Sewage Ingress | Coastal Sediment | Identified a human-specific Bacteroides OTU correlating (R²=0.87) with chemical tracers. | Low microbial biomass led to high stochasticity in replicates below 0.1g sediment. | V4-V5 |
| EuroMST Consortium (2024): Multi-source | Agricultural Runoff | Developed a curated marker database discriminating 6 animal sources with 85% average precision. | Marker abundance dropped below detection after 48 hrs in saturated soils, limiting temporal tracking. | V4 |
Detailed Experimental Protocols
Protocol 2.1: Standardized Water Sample Processing for Low-Biomass MST (Adapted from Chen & Kumar, 2024)
- Objective: To concentrate microbial cells and extract high-quality genomic DNA from water samples for 16S rRNA gene sequencing.
- Materials: Sterile filtration manifold, 0.22µm polycarbonate membranes, PowerWater DNA Isolation Kit (Qiagen), bead-beating tubes, thermal shaker.
- Procedure:
- Filter 500mL to 1L of water sample through a 0.22µm membrane under gentle vacuum (<5 inHg).
- Aseptically fold the membrane and transfer it to a PowerWater bead tube using sterile forceps.
- Add PW1 solution and incubate at 65°C for 10 minutes.
- Perform bead-beating on a thermal shaker at 30 Hz for 5 minutes at 60°C.
- Centrifuge and transfer supernatant to a clean tube.
- Follow the manufacturer's protocol for subsequent binding, washing (PW2 & PW3 buffers), and elution (EB buffer) steps.
- Elute DNA in 50µL of EB buffer. Quantify using a fluorometric assay (e.g., Qubit dsDNA HS Assay).
Protocol 2.2: Bioinformatic Workflow for Source Marker Identification (Adapted from EuroMST Consortium, 2024)
- Objective: To process raw 16S sequences and identify host-specific taxonomic markers.
- Materials: Raw FASTQ files, QIIME2 (v2024.5), SILVA 138 reference database, custom R scripts.
- Procedure:
- Demultiplex & Quality Control: Import paired-end reads into QIIME2. Denoise with DADA2 (--p-trunc-len-f 250 --p-trunc-len-r 200 --p-max-ee-f 2 --p-max-ee-r 3) to generate amplicon sequence variants (ASVs).
- Taxonomy Assignment: Assign taxonomy to ASVs using a pre-trained naive Bayes classifier on the SILVA 138 99% OTUs database for the relevant hypervariable region (e.g., V4).
- Source Filtering: Using metadata, separate sequences from known source samples (e.g., pure fecal samples) to create a source library.
- Marker Analysis: Apply the
q2-source-trackerplugin or execute a custom R script using theFEASTpackage to perform differential abundance analysis (e.g., LEfSe) between source groups. Identify ASVs with >10x enrichment in one source and present in >80% of its replicates. - Validation: Validate candidate markers on an independent set of blinded environmental samples.
Visualizations
Title: 16S rRNA Gene Sequencing MST Core Workflow
Title: Key Limitations in 16S MST & Their Causes
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for 16S rRNA-Based MST Experiments
| Item | Function in MST | Example Product/Brand |
|---|---|---|
| Environmental DNA Isolation Kit | Optimized for efficient lysis of diverse fecal/environmental microbes and removal of PCR inhibitors (humics, organics). | DNeasy PowerSoil Pro Kit (Qiagen), FastDNA Spin Kit for Soil (MP Biomedicals) |
| High-Fidelity PCR Polymerase | Accurate amplification of the target 16S hypervariable region with low error rates to ensure faithful ASV generation. | Q5 Hot Start High-Fidelity DNA Polymerase (NEB), KAPA HiFi HotStart ReadyMix (Roche) |
| Dual-Indexed Primers (16S) | Allows multiplexed sequencing of hundreds of samples with unique barcodes to demultiplex post-run. | 16S V4 Primer Set (515F/806R) with Nextera-style indices (Illumina) |
| Quantitative DNA Standard | For precise quantification of low-concentration environmental DNA, more accurate than absorbance (A260). | Qubit dsDNA HS Assay Kit (Thermo Fisher) |
| Mock Microbial Community | A defined mix of genomic DNA from known species; used as a positive control to assess sequencing run accuracy and bias. | ZymoBIOMICS Microbial Community Standard (Zymo Research) |
| Bioinformatic Pipeline Software | Containerized, reproducible environment for processing raw sequences through quality filtering, ASV calling, and taxonomy. | QIIME 2 Core Distribution, DADA2 R Package |
Integrating 16S Data with Chemical and Physical Markers for Multi-Method MST
1. Application Notes
Within the broader thesis on advancing Microbial Source Tracking (MST) using 16S rRNA gene sequencing, integrating this genetic data with chemical and physical markers represents a critical evolution towards robust, multi-method frameworks. This approach mitigates the limitations of any single method, enhancing the resolution and confidence of fecal pollution source identification in environmental waters.
Key Rationale for Integration:
- Complementary Strengths: 16S sequencing provides high-resolution, library-independent microbial community fingerprints but can be influenced by environmental decay and non-fecal sources. Chemical markers (e.g., pharmaceuticals, fecal stanols) offer source-specific chemical tracers with different degradation kinetics. Physical markers (e.g., water fluorescence, turbidity) provide real-time, bulk water quality indicators.
- Increased Discriminatory Power: Combined data matrices can distinguish between human, ruminant, avian, and other animal sources with greater accuracy than unimodal approaches.
- Temporal and Spatial Dynamics: Multi-parameter tracking allows for the differentiation of recent vs. aged contamination and the identification of multiple concurrent pollution sources.
Quantitative Performance Summary of Integrated Markers:
Table 1: Comparison of MST Marker Classes and Their Integration Value
| Marker Class | Example Targets | Key Strength | Key Limitation | Role in Integrated Framework |
|---|---|---|---|---|
| 16S Genetic | Bacteroides, Lachnospiraceae, host-specific assays | High source specificity, library-independent | DNA persistence ≠ cell viability, PCR inhibition | Provides primary source fingerprint. |
| Chemical | Caffeine, acetaminophen, coprostanol, optical brighteners | Human-specific potential, quantitative | Affected by wastewater treatment, sorption | Confirms human/ruminant sources, indicates wastewater input. |
| Physical | Fluorescence (tryptophan, humic-like), turbidity, conductivity | Real-time, high-frequency measurement | Non-specific, influenced by non-fecal sources | Triggers targeted sampling, indicates pollution events. |
2. Experimental Protocols
Protocol 1: Integrated Water Sample Processing for Multi-Method MST
Objective: To concurrently prepare a single water sample for 16S rRNA gene sequencing, chemical marker analysis (via LC-MS/MS), and physical marker measurement.
Materials:
- Research Reagent Solutions & Essential Materials (See Toolkit Table).
- Sterile, sample-rinsed 1L Nalgene bottles.
- Peristaltic pump or manual vacuum system with 0.22µm pore-size sterivex filters (for microbial biomass).
- Solid Phase Extraction (SPE) apparatus and HLB cartridges (for chemicals).
- In-situ sonde or benchtop fluorometer/spectrophotometer.
Procedure:
- In-situ Physical Marker Measurement: At the sampling site, measure and record parameters (e.g., fluorescence at tryptophan/excitation 280 nm/emission 350 nm, turbidity (NTU), specific conductivity) using a calibrated sonde.
- Sample Collection: Collect 1L of water in a sterile bottle. Process within 6 hours.
- Filtration for Microbial DNA: a. Aseptically filter 500-1000 mL of water through a 0.22µm Sterivex filter to capture bacterial biomass. b. Using sterile forceps, place the filter membrane into a PowerBead tube from the DNeasy PowerWater Kit. Proceed with lysis and DNA extraction per kit protocol. Elute in 50-100 µL of EB buffer. Store at -80°C until 16S library preparation.
- Solid Phase Extraction (SPE) for Chemical Markers: a. Pass the remaining filtrate (or a separate 500mL aliquot) through a preconditioned (methanol, then ultrapure water) 200mg Oasis HLB SPE cartridge at a flow rate of 5-10 mL/min. b. Dry the cartridge under vacuum for 30 minutes. Elute compounds with 2 x 5 mL of methanol into a glass vial. c. Evaporate the eluent to dryness under a gentle stream of nitrogen. Reconstitute in 200 µL of methanol:water (1:1, v/v) for LC-MS/MS analysis.
- Chemical Analysis (LC-MS/MS): Analyze reconstituted samples using a targeted multiple reaction monitoring (MRM) method for a panel of chemical markers (e.g., caffeine, sulfamethoxazole, coprostanol). Use isotope-labeled internal standards for quantification.
Protocol 2: Data Integration and Statistical Workflow
Objective: To combine 16S, chemical, and physical datasets for a unified source attribution.
Procedure:
- Data Normalization: Independently normalize each dataset. For 16S: convert sequence counts to relative abundance or use centered log-ratio (CLR) transformation. For chemicals: normalize to ng/L. For physical: use z-scores.
- Feature Selection: For 16S data, identify the 50-100 most abundant amplicon sequence variants (ASVs) or select known host-associated taxa. For chemical data, select markers above detection limit and with known source association.
- Data Fusion: Combine selected features from all three modalities into a single sample x feature matrix.
- Multivariate Analysis: Perform supervised (e.g., Random Forest, sPLS-DA) and unsupervised (e.g., PCoA based on Bray-Curtis or Aitchison distance) analyses on the fused matrix. Use known source samples (e.g., sewage, septic, animal waste) as a training set.
- Validation: Apply the trained model to unknown environmental samples. Calculate probability assignments and consensus calls from the integrated data.
3. The Scientist's Toolkit
Table 2: Key Research Reagent Solutions & Essential Materials
| Item | Function in Integrated MST |
|---|---|
| DNeasy PowerWater Kit (Qiagen) | Extracts high-quality microbial genomic DNA from environmental water filters, critical for downstream 16S sequencing. |
| Oasis HLB SPE Cartridges (Waters) | Broad-spectrum extraction of diverse chemical markers (acidic, basic, neutral) from large water volumes for concentration. |
| ZymoBIOMICS Microbial Community Standard | A defined mock microbial community used as a positive control and for benchmarking 16S sequencing run performance. |
| Isotope-Labeled Internal Standards (e.g., 13C-caffeine, d4-sulfamethoxazole) | Added prior to chemical extraction to correct for matrix effects and losses during sample preparation for LC-MS/MS. |
| QIIME 2 or DADA2 Pipeline | Open-source bioinformatics platforms for processing raw 16S rRNA sequence data into amplicon sequence variants (ASVs). |
| In-situ Fluorescence/Turbidity Sonde (e.g., YSI EXO) | Provides real-time, concurrent measurements of physical marker parameters at the time of sample collection. |
| MiSeq Reagent Kit v3 (600-cycle) (Illumina) | Standard chemistry for paired-end 300bp sequencing of the 16S rRNA gene V4 region, providing sufficient depth and read length. |
4. Visualizations
Integrated MST Workflow from Sample to Result
Multi-Method Data Fusion Logic
Conclusion
16S rRNA gene sequencing remains a powerful, accessible, and high-throughput cornerstone for Microbial Source Tracking, providing invaluable insights into microbial community composition and contamination sources in biomedical research. While foundational and methodological advancements have standardized its application, researchers must navigate its limitations in resolution and potential biases through rigorous optimization and troubleshooting. The future of MST lies not in relying on a single method, but in the strategic integration of 16S data with complementary techniques like qPCR for specific targets and shotgun metagenomics for strain-level tracking and functional insight. For drug development and clinical settings, this evolving multi-marker approach is crucial for ensuring sterile manufacturing processes, validating cleaning protocols, and ultimately safeguarding product and patient safety. Continued development of curated, host-associated reference databases and standardized bioinformatic pipelines will further solidify the role of 16S rRNA sequencing as an indispensable tool in the microbial investigator's arsenal.