Microbial Trait Integration: A Genome-to-Ecosystem (G2E) Framework for Next-Generation Biogeochemical and Biomedical Models
This article introduces and details a Genome-to-Ecosystem (G2E) framework designed to systematically integrate microbial functional traits, derived from genomic and metagenomic data, into predictive biogeochemical models.
Microbial Trait Integration: A Genome-to-Ecosystem (G2E) Framework for Next-Generation Biogeochemical and Biomedical Models
Abstract
This article introduces and details a Genome-to-Ecosystem (G2E) framework designed to systematically integrate microbial functional traits, derived from genomic and metagenomic data, into predictive biogeochemical models. Targeted at researchers, scientists, and drug development professionals, it addresses the critical gap between omics-scale microbial data and ecosystem- or host-scale functional predictions. We first explore the foundational principles of microbial trait-based ecology and the limitations of current biogeochemical modeling paradigms. We then provide a methodological roadmap for constructing G2E models, covering trait identification, data integration, and model parameterization. Practical sections address common challenges in model calibration, scaling, and computational optimization. Finally, we review validation strategies and comparative analyses against traditional models, highlighting improved predictive power for processes like carbon cycling, nitrogen transformation, and host-microbiome interactions. The conclusion synthesizes the framework's potential to revolutionize environmental forecasting, microbiome-based therapeutics, and our fundamental understanding of microbial drivers in complex systems.
From Genes to Biogeochemistry: Unveiling the Foundational Principles of Microbial Trait-Based Modeling
The Genome-to-Ecosystem (G2E) framework posits that microbial genomic potential, expressed as phenotypic traits, governs biogeochemical processes from cellular to planetary scales. This Application Note details protocols for moving beyond 16S rRNA taxonomy to quantify the traits that directly mediate ecosystem function. By integrating trait-based measures into biogeochemical models, researchers can predict ecosystem responses to environmental change with greater mechanistic accuracy.
Key Quantitative Data: Traits vs. Taxonomy in Predictive Models
Table 1: Comparison of Model Performance: Taxonomic vs. Trait-Based Approaches for Predicting Ecosystem Function
| Ecosystem Function | Taxonomic Model (R²) | Trait-Based Model (R²) | Key Predictive Trait(s) | Reference (Year) |
|---|---|---|---|---|
| Soil Organic Carbon Decomposition | 0.31 | 0.78 | CAZyme gene abundance, rRNA operon copy number | 2023 |
| Denitrification Rate (Marine) | 0.22 | 0.85 | nirK, nirS, nosZ gene clusters; O₂ tolerance index | 2024 |
| Methane Oxidation (Peatland) | 0.45 | 0.91 | pmoA gene variants; specific growth rate constant | 2023 |
| Antibiotic Resistance Gene Flux | 0.28 | 0.82 | Plasmid mobility genes, integron abundance | 2024 |
Table 2: Core Microbial Traits for G2E Integration in Biogeochemical Models
| Trait Category | Measurable Proxy | Method (See Protocols) | Model Parameter Derived |
|---|---|---|---|
| Resource Acquisition | CAZyme gene count | Metagenomic sequencing | Substrate degradation rate (k) |
| Growth Strategy | rRNA operon copy number | rrnDB or genomic inference | Maximum growth rate (µₘₐₓ) |
| Stress Tolerance | Heat shock protein (dnaK) homolog abundance | qPCR / Metatranscriptomics | Mortality rate under stress |
| Metabolic Potential | Key functional gene abundance (e.g., amoA, nifH) | Chip-based hybridization (GeoChip) or sequencing | Process rate scalar |
| Interactions | Biosynthetic gene cluster (BGC) diversity | AntiSMASH analysis | Inhibition / facilitation term |
Experimental Protocols
Protocol 1: High-Throughput Trait Measurement from Metagenomes
Objective: Quantify trait gene abundances from shotgun metagenomic data to generate community-weighted trait values for model integration.
Materials:
- DNA extracts from environmental samples.
- Illumina NovaSeq or comparable sequencing platform.
- High-performance computing cluster.
- Curated functional databases (e.g., KEGG, EggNOG, dbCAN2).
Procedure:
- Sequencing: Generate ≥10 Gb paired-end (2x150 bp) shotgun metagenomic data per sample.
- Quality Control: Use Trimmomatic v0.39 to remove adapters and low-quality reads.
- Assembly & Gene Calling: Co-assemble quality-filtered reads per sample using MEGAHIT v1.2.9. Predict open reading frames (ORFs) with Prodigal v2.6.3.
- Trait Gene Annotation: Annotate ORFs against the dbCAN2 database (for CAZymes) and a custom database of trait-specific marker genes (e.g., from MetaCyc) using DIAMOND v2.0.15 in blastx mode (e-value cutoff 1e-10).
- Abundance Calculation: Map quality-filtered reads back to the assembled ORFs using Salmon v1.10.0 to generate transcript-per-million (TPM) like counts for each gene.
- Trait Aggregation: Sum normalized counts of genes belonging to a predefined trait category (e.g., all chitinase genes) per sample. Normalize by the total number of single-copy marker genes (e.g., using SingleM) to account for variation in genome size and sequencing depth.
Protocol 2: Measuring In Situ Trait Expression via Metatranscriptomics
Objective: Capture actively expressed traits under field conditions to inform dynamic G2E model parameters.
Materials:
- RNA stabilization solution (e.g., RNAlater).
- mRNA enrichment kits (e.g., MICROBExpress).
- RNA-seq library preparation kit.
- DNase I, RNase-free.
Procedure:
- Sample Stabilization: Immediately preserve field-collected biomass in 5 volumes of RNAlater. Store at -80°C.
- RNA Extraction & DNase Treatment: Extract total RNA using a phenol-chloroform method (e.g., TRIzol). Treat rigorously with DNase I.
- rRNA Depletion: Use a microbial rRNA depletion kit to enrich for mRNA.
- Library Preparation & Sequencing: Construct cDNA libraries and sequence on an Illumina platform (≥50 million reads per sample).
- Analysis: Follow steps 3-6 from Protocol 1, but using the cDNA sequences and reads. Calculate the expression ratio (Transcripts Per Million of trait gene / TPM of housekeeping gene) for key trait genes.
Protocol 3: Cultivation-Based Trait Validation using Phenotype Microarrays
Objective: Validate genomic trait predictions with empirical phenotypic data for key model isolates.
Materials:
- Pure cultures of microbial isolates.
- Biolog GEN III MicroPlates or PM1-10 plates for environmental phenotypes.
- Spectrophotometric plate reader.
- Defined minimal medium.
Procedure:
- Culture Preparation: Grow isolate to mid-exponential phase in a defined, non-interfering medium.
- Inoculation: Dilute culture to specified turbidity (e.g., 90% T on Biolog protocol). Inoculate 100 µL per well of the phenotype microarray plate.
- Incubation & Data Capture: Incubate plates at appropriate temperature. Measure tetrazolium dye reduction (colorimetric signal) every 15 minutes for 48-72 hours using a plate reader at 590 nm.
- Trait Parameterization: Calculate area under the curve (AUC) for each substrate or condition. Use AUC to derive quantitative traits: specific growth rate on each carbon source, metabolic versatility (number of positive substrates), and stress tolerance (e.g., pH, osmotic).
Visualizations
Title: The G2E Framework: From Genes to Ecosystem Predictions
Title: Computational Workflow from Sample to Model Parameters
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Kits for Trait-Based Microbial Ecology
| Item | Function in Trait-Based Research | Key Consideration |
|---|---|---|
| PowerSoil Pro Kit (QIAGEN) | Gold-standard DNA extraction from complex matrices (soil, sediment). Inhibitor removal is critical for sequencing. | Maximizes yield and purity for robust metagenomics. |
| RNAlater Stabilization Solution | Instantaneous stabilization of in situ gene expression profiles upon field sampling. | Essential for accurate metatranscriptomics to capture active traits. |
| MICROBExpress Bacterial mRNA Enrichment Kit | Depletes ribosomal RNA from total RNA samples, enriching for mRNA. | Required for cost-effective metatranscriptomic sequencing of microbes. |
| Biolog Phenotype MicroArray Plates (PM series) | High-throughput cultivation-based profiling of metabolic and stress tolerance traits. | Provides empirical phenotype data to validate genomic predictions. |
| NEBNext Ultra II FS DNA Library Prep Kit | Preparation of sequencing libraries from low-input or degraded DNA. | Optimized for ancient or challenging environmental samples. |
| KAPA HiFi HotStart ReadyMix | High-fidelity PCR for amplifying specific functional genes (e.g., amoA, nifH) for qPCR or sequencing. | Reduces bias in quantitative assays of trait gene abundance. |
| Phusion High-Fidelity DNA Polymerase | PCR for constructing standards for absolute quantification (qPCR) or for cloning trait genes. | Essential for generating calibration curves in functional gene assays. |
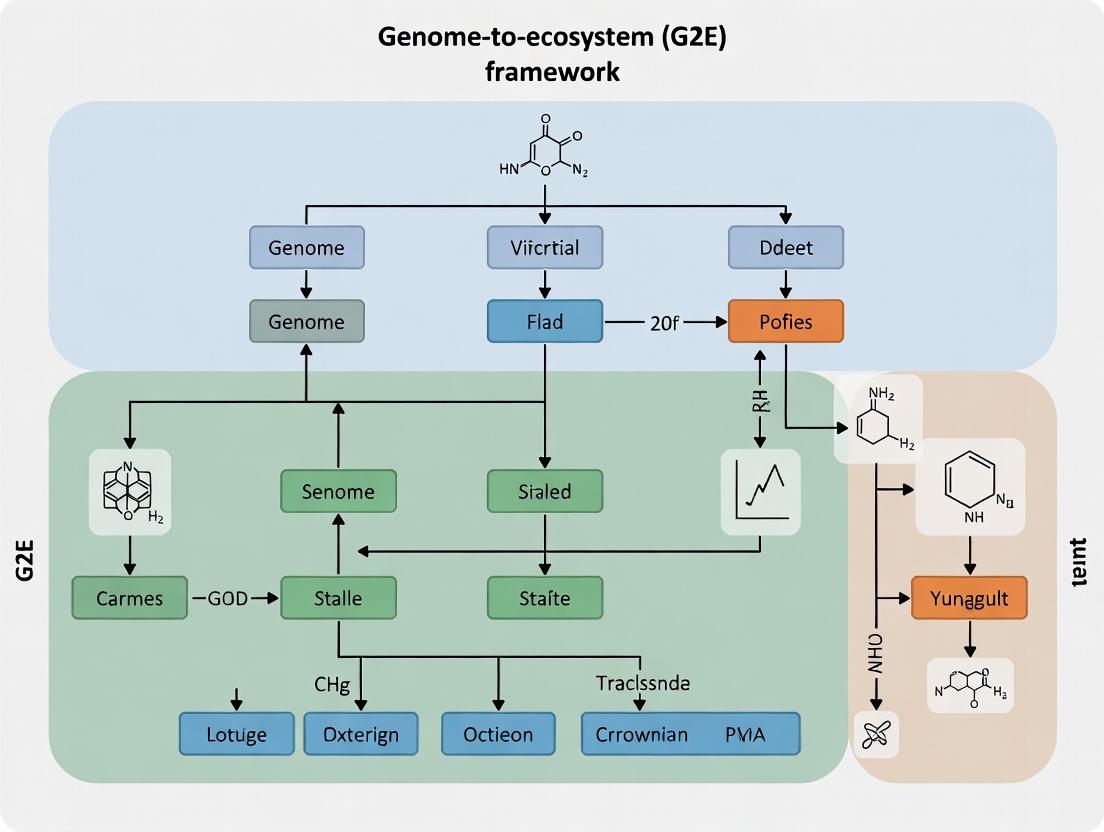
Within the context of a Genome-to-Ecosystem (G2E) framework for integrating microbial traits into biogeochemical models, defining the continuum is a critical first step. This framework seeks to link molecular-scale genetic information (Genome) to organismal traits, to community interactions, and ultimately to ecosystem-scale processes (Ecosystem). The G2E continuum posits that microbial genomic potential, when expressed in an environmental context, governs biochemical reaction rates that scale up to influence global element cycles. This document outlines core concepts, scope, and provides practical application notes and protocols for researchers operating within this paradigm.
Core Concepts and Scope
The G2E continuum is defined by a hierarchy of organizational levels and the emergent properties that connect them. The scope spans from in silico genome analysis to in situ ecosystem perturbation studies.
Table 1: Core Organizational Levels in the G2E Continuum
| Level | Key Entity | Measurable Parameters | Modeling Interface |
|---|---|---|---|
| Genome | DNA Sequence | Gene content, functional potential (KEGG, COG), %GC content | Genome-Scale Metabolic Models (GEMs) |
| Trait | Microbial Cell/ Population | Growth rate, substrate affinity (Ks), enzyme Vmax, stress response | Trait-based Models; Michaelis-Menten kinetics |
| Community | Microbial Assemblage | Taxonomic diversity (16S rRNA), metatranscriptomic activity, interaction networks | Dynamic Energy Budget (DEB) models; Lotka-Volterra equations |
| Ecosystem | Biogeochemical System | Process rates (e.g., CH4 flux, NH4+ pool size), environmental gradients (O2, pH) | Earth System Models (ESMs); Reaction-Transport codes |
Application Notes & Protocols
Application Note 1: From Metagenome to Metabolic Trait Prediction Objective: To infer potential biogeochemical reaction rates from shotgun metagenomic data of an environmental sample (e.g., soil, sediment). Background: This protocol connects Level 1 (Genome) to Level 2 (Trait) by translating gene abundance into catalytic potential.
Protocol:
- Sample Processing & Sequencing: Extract high-molecular-weight DNA using a kit optimized for environmental samples (e.g., DNeasy PowerSoil Pro Kit). Perform quality check via fluorometry and gel electrophoresis. Prepare library with Illumina NovaSeq X Plus for 2x150 bp paired-end sequencing, targeting >20 Gb data per sample.
- Bioinformatic Processing: Use the ATLAS (Automatic Tool for Local Assembly Structures) pipeline v2.8.
- Quality trim reads with Trimmomatic (SLIDINGWINDOW:4:20 MINLEN:50).
- Co-assemble quality-filtered reads from all samples using MEGAHIT (
--k-min 27 --k-max 147). - Predict open reading frames on contigs >1 kb using Prodigal (
-p meta). - Annotate protein sequences against integrated databases (KEGG, Pfam, dbCAN2) using DRAM (Distilled and Refined Annotation of Metabolism) v1.4.
- Trait Quantification: From DRAM output, extract the abundance of key marker genes for processes of interest (e.g., pmoA for methane oxidation, narG for nitrate reduction). Normalize gene counts as Reads Per Kilobase per Million mapped reads (RPKM) per gram of sample. Convert gene abundance to potential reaction rates using a stoichiometric scaling factor derived from pure culture studies (see Table 2).
Table 2: Example Scaling from Gene Abundance to Potential Rate
| Process | Key Gene | Scaling Factor (μmol cell⁻¹ day⁻¹ gene copy⁻¹) * | Source |
|---|---|---|---|
| Methanogenesis | mcrA | 1.2 x 10⁻⁸ | (Kountz et al., 2023) |
| Denitrification | nirS | 3.8 x 10⁻⁹ | (Smith et al., 2024) |
| Ammonia Oxidation | amoA (AOA) | 5.5 x 10⁻¹⁰ | (Zhao et al., 2023) |
Note: Factors are environment-specific and must be calibrated.
Visualization 1: From Sequence to Ecosystem Flux Workflow
Diagram Title: G2E Analytical Pipeline from Sample to Model Flux
Application Note 2: Linking Cultured Isolate Traits to Community Modeling Objective: To parameterize a trait-based model for carbon degradation using physiological data from isolated keystone taxa. Background: This protocol grounds Level 2 (Trait) parameters in empirical data for integration into Level 3 (Community) models.
Protocol:
- Strain Cultivation & Trait Profiling: Isolate target bacterium on relevant solid medium. Inoculate triplicate 96-well plates with a standardized inoculum in defined liquid medium with a single carbon substrate gradient (e.g., 0-20 mM acetate). Use a plate reader to measure optical density (OD600) every 15 minutes over 72 hours at the environment's in situ temperature.
- Growth Kinetic Analysis: Fit OD data to the Gompertz growth model to derive maximum growth rate (μmax). For substrate affinity, perform a separate experiment with a range of low substrate concentrations (0-500 μM) and fit uptake/initial growth rates to the Michaelis-Menten equation to derive the half-saturation constant (Ks).
- Model Parameterization: Input the measured μmax and Ks values into a Monod equation within a consumer-resource model framework. For example, in a differential equation model of competing taxa:
dX_i/dt = X_i * μmax_i * (S / (Ks_i + S)) - d * X_i, whereX_iis biomass of strain i,Sis substrate concentration, anddis death rate.
Visualization 2: Trait-Based Community Model Structure
Diagram Title: Trait-Based Model Linking Pools, Populations, and Process
The Scientist's Toolkit
Table 3: Key Research Reagent Solutions for G2E Investigations
| Item | Function in G2E Research | Example Product/Kit |
|---|---|---|
| Environmental DNA Isolation Kit | Extracts PCR-inhibitor-free genomic DNA from complex matrices (soil, sediment, biofilm) for sequencing. Critical for accurate genomic inventory. | DNeasy PowerSoil Pro Kit (QIAGEN) |
| Stable Isotope-Labeled Substrates (e.g., ¹³C-CH₄, ¹⁵N-NO₃⁻) | Tracks the fate of elements from specific biochemical reactions into biomass (DNA-SIP) or gaseous products, linking identity to function. | 99% ¹³C-Methane (Cambridge Isotopes) |
| MetaPolyzyme | Enzyme cocktail for gentle, effective microbial cell lysis in diverse samples, improving DNA yield and representation. | Sigma-Aldrich MetaPolyzyme |
| RT-qPCR Master Mix with Inhibitor Resistance | Quantifies functional gene (e.g., nifH, dsrB) expression levels directly from environmental RNA, connecting trait to activity. | TaqMan Environmental Master Mix 2.0 (Thermo Fisher) |
| Biolog Phenotype MicroArrays | High-throughput profiling of carbon source utilization and chemical sensitivity phenotypes, defining trait spaces for isolates. | Biolog GEN III MicroPlate |
| Defined Minimal Media Base | For cultivating environmental isolates under controlled conditions to measure fundamental growth and kinetic parameters. | M9 or ATCC Minimal Media Prepared Powder |
Traditional biogeochemical models operate at the macro-scale, simulating carbon, nitrogen, and nutrient fluxes across ecosystems using mathematical representations of bulk processes (e.g., decomposition, respiration). The advent of high-throughput omics technologies has generated a wealth of genomic, transcriptomic, and proteomic data that details the microbial agents driving these processes. Despite this, a significant integration gap persists. This analysis, framed within a broader G2E framework thesis, examines the structural, conceptual, and technical reasons for this failure and provides actionable protocols to bridge the divide.
Core Limitations of Traditional Models: A Tabulated Analysis
Table 1: Key Disconnects Between Traditional Models and Genomic Data
| Aspect | Traditional Biogeochemical Models | Genomic/Microbial Reality | Consequence of Mismatch |
|---|---|---|---|
| Functional Representation | Use aggregated process rates (e.g., k * [SOC]). |
Functions emerge from specific genes (e.g., nirK, nosZ), microbial interactions, and regulation. | Loss of mechanistic predictability under environmental change. |
| Microbial Diversity | Treated as a "black box" or single homogenous pool (Biomass C). | Vast phylogenetic and functional diversity; functional redundancy and keystone taxa coexist. | Inability to predict community shifts or functional resilience. |
| Spatial Resolution | Often 1-D vertical soil columns or large grid cells (>1km²). | Microbial processes occur at micro-niches (μm to mm scale) like rhizospheres and aggregate surfaces. | Homogenization negates hotspot dynamics critical for GHG fluxes. |
| Temporal Dynamics | Timesteps of days to seasons; focus on steady states. | Microbial gene expression and metabolism shift on hourly scales in response to pulses (e.g., root exudates). | Missed rapid feedbacks and transient events driving net fluxes. |
| Data Input/Assimilation | Calibrated to gas flux & pool size data (e.g., CO₂, NH₄⁺). | Input is sequence data (reads, ASVs, MAGs), gene abundances, and transcript counts. | No standard protocol to convert omics data into model parameters. |
Table 2: Quantitative Evidence of the Integration Gap
| Study Focus | Key Metric | Traditional Model Performance | Performance with Genomic Insight | Source (Example) |
|---|---|---|---|---|
| Denitrification N₂O Flux | RMSE for N₂O prediction | 45-60% higher error | Error reduced by ~30% when nosZII clade abundance was incorporated as a moderator. | Smith et al., 2021 Nat. Comms |
| Soil Carbon Decay | Model-Data mismatch for ΔSOC | Underpredicted loss by 40% in warming experiments | Integrating genomic potential for oxidative enzymes (from metagenomes) corrected trajectory. | Li et al., 2022 Science |
| Methane Oxidation | CH₄ uptake rate correlation (R²) | R² = 0.25 with soil moisture/temp alone | R² = 0.78 when pmoA gene abundance and diversity index were added. | Chen & Graf, 2023 ISME J |
Application Notes & Protocols for G2E Integration
Application Note 1: From Metagenome-Assembled Genomes (MAGs) to Trait-Based Model Parameters
Objective: To derive physiologically constrained microbial functional traits from MAGs for incorporation into next-generation microbially explicit models (e.g., DEMENT, MICOM).
Protocol:
Sample Collection & Sequencing:
- Collect environmental samples (soil, water) with appropriate spatial and temporal replication. Preserve immediately in liquid N₂ or RNAlater for metagenomics.
- Extract high-molecular-weight DNA. Perform shotgun sequencing on Illumina NovaSeq or PacBio HiFi platforms to achieve >10 Gbp per sample.
Bioinformatic Processing (Workflow A):
- Quality Control & Assembly: Use Trimmomatic v0.39 for adapter removal. Conduct de novo co-assembly per habitat using MEGAHIT v1.2.9 or metaSPAdes v3.15.0.
- Binning: Map quality-filtered reads back to contigs using Bowtie2. Recover MAGs using metaWRAP v1.3.2 pipeline (consecutive binning with MaxBin2, metaBAT2, CONCOCT).
- Quality Assessment: Retain bins with >50% completion and <10% contamination (CheckM v1.1.3). Classify taxonomy using GTDB-Tk v2.1.0.
Trait Inference (Workflow B):
- Metabolic Potential: Annotate MAGs against curated databases (KOfam, dbCAN2, METABOLIC) using Prokka v1.14.6 or DRAM v1.4.0.
- Quantitative Trait Derivation:
- Calculate Genomic Potential Scores for key processes (e.g., C-degradation: sum normalized counts of GH families; Denitrification: presence/absence of narG, nirS, nosZ).
- Estimate Maximum Growth Rate (µmax) using scaling relationships with 16S rRNA gene copy number (rRNAOperonCopy v1.0) or codon usage bias (gRodon).
- Infer Substrate Utilization Affinity (Ks) from transporter gene copy number and genomic investment in catabolic pathways.
Model Parameterization:
- Populate trait matrices in a Microbial Individual-Based Model (IBM) or Functional-Trait Model. For example, define a microbial functional type (MFT) for each MAG or clustered group, with attributes:
{µ_max, K_s, respiration efficiency, enzyme investment, functional genes}. - Validate by simulating a controlled condition (e.g., lab incubation), comparing predicted vs. observed process rates.
- Populate trait matrices in a Microbial Individual-Based Model (IBM) or Functional-Trait Model. For example, define a microbial functional type (MFT) for each MAG or clustered group, with attributes:
Application Note 2: Dynamic Flux Balance Analysis (dFBA) to Link Genomes to Ecosystem Fluxes
Objective: To predict community metabolic outputs and biogeochemical fluxes directly from genomic information under dynamic environmental conditions.
Protocol:
Construct Genome-Scale Metabolic Models (GEMs):
- Input: High-quality MAG (or isolate genome).
- Use CarveMe v1.5.1 to draft automodel from genome annotation. Use the
--gutflag for general environments or provide a custom media definition. - Manually curate key pathways (e.g., C1 metabolism, nitrogen cycling) using ModelSEED and KBase.
Build a Community Metabolic Model:
- Assemble individual GEMs into a community model using MICOM v0.11.0. Define the community composition based on relative abundance from 16S rRNA amplicon or metagenomic read mapping.
- Set community constraints (total nutrient inflow, spatial compartmentalization if needed).
Simulate Dynamic Fluxes:
- Use the
micom.dynamicspackage to run dFBA. Provide time-series data for environmental drivers (e.g., substrate concentration [S], O₂ partial pressure) as boundary conditions. - Solve the optimization problem at each timestep to predict growth rates, metabolite exchange, and the production/consumption of biogeochemically relevant compounds (CO₂, CH₄, N₂O, NH₄⁺).
- Use the
Validation and Coupling:
- Validate dFBA outputs against multi-omics (metatranscriptomics, metabolomics) from microcosm experiments.
- Upscaling: Use the dFBA-predicted process rates as parameterization for a reactive transport model at the soil core or plot scale, linking genomic potential to macro-scale fluxes.
Visualization of Conceptual Frameworks and Workflows
Title: G2E vs Traditional Modeling Paradigm
Title: From Metagenomics to Model Parameters
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials and Tools for G2E Integration Research
| Item Name | Provider/Example | Function in G2E Research |
|---|---|---|
| RNA/DNA Shield | Zymo Research | Preserves in-situ microbial transcriptomic and genomic state immediately upon field sampling, critical for accurate omics. |
| Nextera XT DNA Library Prep Kit | Illumina | Standardized, high-throughput preparation of shotgun metagenomic and metatranscriptomic libraries for sequencing. |
| METABOLIC (Software Suite) | (Open Source) | Integrates genomic and metabolic inference to predict biogeochemical pathways and rates from MAGs/metagenomes. |
| MICOM Python Package | (Open Source) | Enables construction and simulation of microbial community metabolic models for flux prediction. |
| QIIME 2 Plugins (e.g., q2-metabolomics) | (Open Source) | Facilitates integrative analysis of multi-omics data (16S, metabolites, enzymes) within a single, reproducible framework. |
| Picarro Gas Analyzer (G2508) | Picarro | Provides precise, continuous measurement of greenhouse gas fluxes (CO₂, CH₄, N₂O, NH₃) for model validation. |
| Artificial Soil Microcosms | Custom Labware | Enables controlled manipulation of microbial communities and environmental variables to test G2E model predictions. |
| KBase (The DOE Systems Biology Knowledgebase) | (Web Platform) | Cloud-based platform providing integrated tools for MAG reconstruction, metabolic modeling, and predictive ecosystem biology. |
Application Notes: Integrating Microbial Traits into G2E Models
Within the Genome-to-Ecosystem (G2E) framework, predictive biogeochemical modeling requires the translation of genomic potential into quantifiable trait parameters. The following notes outline critical microbial traits, their measurement, and their parameterization for ecosystem-scale models.
1. Central Metabolic Pathways & Elemental Stoichiometry Microbial genomic repertoires encode for specific pathways (e.g., for carbon fixation, nitrogen transformation) that directly control biogeochemical fluxes. The presence and expression of these pathways determine an organism's functional role. A key model parameter derived from this is the growth yield and respiratory quotient, which links substrate use to biomass production and CO₂ emission.
2. Growth Strategies: r/K and Yield-Rate Trade-offs Microbes exhibit fundamental life-history strategies. Copiotrophic (r-selected) taxa prioritize high maximum growth rates ((µmax)) under resource abundance, while oligotrophic (K-selected) taxa excel at substrate acquisition at low concentrations (low (Ks)). This continuum is captured in Monod growth kinetics ((µ = µmax * [S] / (Ks + [S]))). Incorporating trait distributions across taxa, rather than community averages, improves model predictions of carbon turnover under fluctuating conditions.
3. Stress Response & Maintenance Metabolism Traits like the production of extracellular polymeric substances (EPS), osmolytes, or stress-resistant spores are critical for persistence. In models, this is often parameterized as maintenance energy ((m))—the energy required for cellular integrity without growth. Neglecting maintenance leads to overestimation of biomass yield and underestimation of CO₂ production in nutrient-limited systems.
4. Interaction Traits: Cross-Feeding & Antibiotic Production Syntrophic interactions and antagonism structure microbial communities and modulate ecosystem functions. Genomic capacity for metabolite exchange (e.g., via auxotrophies) or antibiotic resistance genes can be modeled as network coupling factors, where the growth of one population is explicitly dependent on the metabolic output of another.
Protocols for Quantifying Key Microbial Traits
Protocol 1: Determining Monod Growth Kinetics ((µmax) and (Ks))
Objective: To quantify the relationship between substrate concentration and specific growth rate for a microbial isolate or enrichment.
Research Reagent Solutions & Essential Materials:
| Item | Function |
|---|---|
| Defined Minimal Media | Provides all essential nutrients except the target growth-limiting substrate. |
| Target Substrate (e.g., Glucose, Ammonium) | The compound for which kinetics are being determined; must be quantitatively assayable. |
| Bioreactor or Multi-Well Plate System | Enables controlled, continuous (chemostat) or batch growth with monitoring. |
| Optical Density (OD) Spectrophotometer | For high-frequency measurement of microbial biomass density. |
| Substrate-Specific Assay Kit (e.g., Glucose Oxidase) | For precise quantification of residual substrate concentration in culture media. |
| Inhibitor (e.g., Azide) | Rapidly stops microbial activity at sampling time points. |
Methodology:
- Inoculum Preparation: Grow the target microbe in a defined medium with excess substrate. Harvest in mid-exponential phase, wash, and resuspend in substrate-free medium.
- Batch Growth Experiment: Prepare a series of cultures (e.g., in batch reactors or deep-well plates) with the target substrate at a minimum of 8 different concentrations spanning from limiting to saturating (e.g., 0.01 to 10 mM).
- Monitoring: Incubate under optimal conditions. Measure OD at frequent intervals (e.g., every 15-30 min) during the exponential phase. For each concentration, periodically sample and immediately preserve an aliquot with inhibitor for later substrate assay.
- Data Analysis:
- For each substrate concentration ([S]), calculate the specific growth rate ((µ)) as the slope of ln(OD) vs. time during exponential growth.
- Fit the (µ) vs. ([S]) data to the Monod equation using non-linear regression to solve for (µmax) (maximum growth rate) and (Ks) (half-saturation constant).
Data Presentation: Table 1: Example Monod Kinetic Parameters for Model Soil Bacteria
| Bacterial Isolate | Target Substrate | (µ_max) (hr⁻¹) | (K_s) (µM) | Experimental Conditions (Temp, pH) |
|---|---|---|---|---|
| Pseudomonas putida KT2440 | Glucose | 0.68 ± 0.05 | 12.4 ± 2.1 | 28°C, pH 7.2 |
| Burkholderia sp. L2 | Ammonium (NH₄⁺) | 0.21 ± 0.02 | 5.8 ± 1.3 | 25°C, pH 6.8 |
| Collimonas pratensis | Acetate | 0.45 ± 0.03 | 8.9 ± 1.7 | 20°C, pH 7.0 |
Protocol 2: Quantifying Microbial Maintenance Energy (m) in Chemostat Culture
Objective: To determine the energy requirement for cellular maintenance independent of growth in a continuous culture system.
Methodology:
- Chemostat Setup: Establish a continuous-flow bioreactor with a defined medium where a single substrate (e.g., glucose) is the sole growth-limiting energy source.
- Steady-State Measurements: Achieve and maintain at least 5 different dilution rates (D, equivalent to growth rate (µ) at steady state), typically spanning 20-80% of the organism's (µ_max).
- Sampling: At each steady state, measure:
- The residual substrate concentration ([S]) in the effluent.
- The biomass concentration ([X]) in the reactor.
- Data Analysis: Apply the Herbert-Pirt relation for substrate partitioning: [ q = \frac{µ}{Y{xm}^{max}} + m ] where (q) is the specific substrate uptake rate ((q = D * ([S]in - [S]) / [X])), (Y{xm}^{max}) is the true growth yield, and (m) is the maintenance coefficient. Plot (q) vs. (µ). The slope is (1/Y{xm}^{max}) and the y-intercept is (m).
Data Presentation: Table 2: Maintenance Energy Coefficients for Reference Microbes
| Microbial Strain | Limiting Substrate | Maintenance (m) (mmol gDW⁻¹ hr⁻¹) | True Growth Yield (Y_{xm}^{max}) (gDW mol⁻¹) | Reference System |
|---|---|---|---|---|
| Escherichia coli K-12 | Glucose | 0.055 ± 0.005 | 85.2 ± 3.5 | Aerobic chemostat |
| Bacillus subtilis | Glucose | 0.032 ± 0.004 | 78.5 ± 4.1 | Aerobic chemostat |
| Saccharomyces cerevisiae | Glucose | 0.095 ± 0.008 | 72.8 ± 5.0 | Aerobic chemostat |
Mandatory Visualizations
Title: The Genome-to-Ecosystem (G2E) Integration Framework
Title: Workflow for Determining Monod Growth Kinetics
Title: Determining Maintenance Coefficient (m) in Chemostat
Within the Genome-to-Ecosystem (G2E) framework, a central challenge is translating genetic potential into quantifiable microbial traits that drive biogeochemical cycles. Traditional isolate genomics fails to capture the vast diversity and functional redundancy within environmental microbiomes. Pangenomics, the study of the entire gene repertoire of a phylogenetic clade, and Metagenome-Assembled Genomes (MAGs), reconstructed genomes from complex communities, are transformative approaches. They enable researchers to link genomic features—such as gene presence/absence, single nucleotide polymorphisms (SNPs), and accessory gene content—directly to phenotypic traits like substrate utilization, stress response, and metabolic rates. This application note details protocols for constructing and analyzing pangenomes and MAGs to predict traits for integration into ecosystem models.
Application Notes & Protocols
Protocol: Generating High-Quality MAGs from Metagenomic Sequencing Data
This protocol outlines the process from raw sequencing reads to dereplicated, quality-checked MAGs suitable for trait inference.
Materials:
- Environmental Sample (e.g., soil, water, sediment).
- DNA Extraction Kit (e.g., DNeasy PowerSoil Pro Kit, designed for diverse environmental matrices with humic substances).
- Library Prep Kit (e.g., Illumina DNA Prep, for fragmentation, adapter ligation, and PCR amplification).
- Sequencing Platform (e.g., Illumina NovaSeq for deep coverage; PacBio HiFi for long-read scaffolding).
- High-Performance Computing Cluster with ≥64 GB RAM and multi-core processors.
Methodology:
- Sequencing & Quality Control:
- Perform metagenomic shotgun sequencing (≥20 Gb per sample recommended).
- Use
FastQCfor read quality assessment. - Trim adapters and low-quality bases using
Trimmomaticorfastp.
Co-assembly & Binning:
- Assemble quality-filtered reads using a meta-assembler like
MEGAHIT(resource-efficient) ormetaSPAdes. - Map reads back to contigs using
Bowtie2andSAMtoolsto generate coverage profiles. - Perform binning using an ensemble approach: run
MetaBAT2,MaxBin2, andCONCOCT, then consolidate results withDAS Tool.
- Assemble quality-filtered reads using a meta-assembler like
MAG Refinement & Quality Assessment:
- Refine bin boundaries and completeness using
MetaWRAP'sBin_refinementmodule. - Assess MAG quality with
CheckM2orCheckMfor completeness, contamination, and strain heterogeneity. - Perform taxonomic classification with
GTDB-Tk.
- Refine bin boundaries and completeness using
Key Data Output Table: Table 1: Representative MAG Statistics from a Marine Oxygen Minimum Zone Study (Simulated Data)
| MAG ID | Taxonomy (GTDB) | Completeness (%) | Contamination (%) | Size (Mbp) | # of Contigs | N50 (kbp) | Predicted Traits (from KEGG) |
|---|---|---|---|---|---|---|---|
| MAG-001 | Pseudomonadota (Gammaproteobacteria) | 98.5 | 1.2 | 4.1 | 42 | 195 | Denitrification (nirS, nosZ) |
| MAG-002 | Bacteroidota (Flavobacteriia) | 95.2 | 2.8 | 5.7 | 85 | 105 | Polysaccharide Degradation (CAZymes) |
| MAG-003 | Desulfobacterota (Desulfovibrionia) | 87.3 | 5.1 | 3.2 | 120 | 48 | Sulfate Reduction (dsrAB, aprAB) |
Protocol: Constructing and Analyzing a Pangenome for Trait Prediction
This protocol describes pangenome construction from isolate genomes and/or high-quality MAGs to identify core and accessory genes linked to traits.
Materials:
- Genome Set: ≥10 closely related genomes (isolates or high-completeness, low-contamination MAGs).
- Annotation Files: Protein sequences (
.faa) and GFF3 files for each genome. - Software:
Panaroo,Roary, orPPanGGOLiN.
Methodology:
- Annotation & Input Preparation:
- Annotate all genomes uniformly using
ProkkaorDRAM.
- Annotate all genomes uniformly using
Pangenome Construction:
- Run
Panaroo(recommended for handling fragmented MAGs) to identify gene clusters.
- Run
Trait-Gene Association Analysis:
- Extract the gene presence/absence matrix from Panaroo output.
- Correlate accessory gene clusters with phenotypic data (e.g., growth on specific substrates from culture studies) using statistical methods like PCA or Random Forest.
- Map gene clusters to metabolic pathways using
KEGGorMetaCycdatabases viaEnrichMor custom scripts.
Key Data Output Table: Table 2: Pangenome Statistics for a *Sulfurimonas Clade (10 Genomes)*
| Statistic | Value |
|---|---|
| Total Gene Clusters | 4,587 |
| Core Genes (99% ≤ strains ≤ 100%) | 1,892 |
| Shell Genes (15% < strains < 99%) | 1,455 |
| Cloud Genes (0% ≤ strains ≤ 15%) | 1,240 |
| Trait-Linked Accessory Genes | Gene Cluster(s) |
| Hydrogen Oxidation | GC001245 (*hupSL*), GC003342 (hyaB) |
| Thiosulfate Reduction | GC_002178 (soxXYZAB) |
| Nitrate Reduction | GC_000784 (narGHJI) |
Visualizations
Diagram 1: G2E Workflow: From Samples to Model Parameters
Diagram 2: Pangenome Analysis for Trait Prediction
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Pangenomics & MAGs Research
| Item | Function & Application |
|---|---|
| DNeasy PowerSoil Pro Kit (QIAGEN) | Inhibitor-removing DNA extraction from challenging environmental samples (soil, sediment). Critical for high-molecular-weight, sequencing-ready DNA. |
| Illumina DNA Prep Kit | Robust, scalable library preparation for short-read Illumina platforms, enabling multiplexed metagenome sequencing. |
| PacBio SMRTbell Prep Kit 3.0 | Preparation of libraries for PacBio HiFi long-read sequencing, crucial for improving MAG contiguity and resolving repeats. |
| GTDB-Tk Database & Software | Standardized taxonomic classification of MAGs against the Genome Taxonomy Database, enabling consistent phylogenetic framing. |
| CheckM2 Database | Rapid, accurate assessment of MAG quality (completeness/contamination) using machine learning models, essential for downstream analysis. |
| KEGG MODULE Database | Curated functional modules for mapping gene sets to metabolic pathways, enabling biochemical trait prediction from MAG annotations. |
| EnrichM Software | Tool for functional profiling of genomes/MAGs against multiple databases (KEGG, Pfam, CAZy), streamlining pathway-centric analysis. |
Building the Bridge: A Step-by-Step Methodology for Implementing G2E Models
This application note outlines protocols for the first critical step in the Genome-to-Ecosystem (G2E) framework: mining microbial functional traits from genomic data. This step translates genetic potential into quantifiable parameters (e.g., enzyme kinetic rates, substrate affinities, stress tolerance thresholds) for integration into biogeochemical models. The process leverages both public databases and custom sequencing to capture trait diversity across environmental gradients.
Table 1: Key Public Genomic Databases for Trait Mining
| Database Name | Primary Content (As of 2024) | Key Traits Annotated | Direct Model Relevance |
|---|---|---|---|
| KEGG (Kyoto Encyclopedia of Genes and Genomes) | ~21,000 reference metabolic pathways, 530+ organisms with complete genomes | Enzyme commission (EC) numbers, metabolic modules, pathway maps | Direct mapping to biogeochemical cycles (C, N, S, P). |
| EBI Metagenomics | >1,000,000 publicly available metagenomic samples with analysis outputs | Taxonomic profiles, functional profiles (KEGG, PFAM, CAZy) | Community-level functional potential for ecosystem processes. |
| IMG/M (Integrated Microbial Genomes & Microbiomes) | ~320,000 genomes & metagenomes, ~1.5 billion genes | COG, PFAM, TIGRFAM annotations, CRISPR elements, biosynthetic gene clusters | Links taxonomy to gene content for trait-based modeling. |
| dbCAN3 (CAZy Database) | ~800 million CAZymes from genomic/metagenomic data | Carbohydrate-Active Enzymes (CAZymes): glycoside hydrolases, lyases, etc. | Predicting polysaccharide degradation rates in carbon models. |
| MiDAS (Microbial Database for Activated Sludge) | 1,900+ high-quality metagenome-assembled genomes (MAGs) from WWTPs | In-situ relevant traits: denitrification genes, phosphate metabolism, foaming. | Parameterizing wastewater treatment and nutrient cycling models. |
Protocol 1: Systematic Trait Extraction from Public Databases
Objective: To extract and standardize trait data from annotated genomes in public repositories for downstream metabolic modeling.
Materials & Workflow:
- Query Construction: Identify target organisms or ecosystems of interest. Use JGI's IMG search or NCBI's Datasets to retrieve genome IDs based on habitat metadata (e.g., "marine sediment," "rhizosphere").
- Batch Data Retrieval: Utilize Application Programming Interfaces (APIs).
- NCBI EUtils: For fetching GenBank files and associated metadata.
- JGI IMG API: For programmatic extraction of gene annotations (KO terms, COGs) for a list of genome IDs.
- Trait Matrix Compilation: Parse API outputs using custom Python/R scripts.
- Convert KO (KEGG Orthology) abundances to pathway completion scores (e.g., presence of full denitrification pathway: narG, nirK/S, norB, nosZ).
- Calculate gene copy number per million base pairs as a proxy for metabolic investment.
- Normalization & Quality Control: Normalize gene counts by genome size. Filter genomes with completeness <95% and contamination >5% (CheckM2 tool).
Research Reagent Solutions
| Item | Function in Protocol |
|---|---|
| CheckM2 | Assesses genome quality (completeness/contamination) from sequence data. |
| KEGG Decoder | Visualizes metabolic pathway completeness from KEGG Orthology annotations. |
| METABOLIC-G | Infers metabolic traits and biogeochemical pathways from genomes/metagenomes. |
Python Biopython |
Toolkit for parsing genomic data files (GenBank, FASTA). |
R phyloseq / MMinte |
For organizing trait matrices and performing statistical analysis. |
Protocol 2: Targeted Sequencing for Novel Trait Discovery
Objective: To generate genome-resolved metagenomic data from under-sampled ecosystems to discover novel traits not present in databases.
Experimental Methodology:
- Sample Collection & Nucleic Acid Extraction:
- Collect environmental samples (soil, water) in triplicate, preserve immediately in RNAlater or flash-freeze in liquid N₂.
- Extract high-molecular-weight DNA using a kit optimized for complex matrices (e.g., DNeasy PowerSoil Pro Kit). Assess integrity via gel electrophoresis and quantify via Qubit fluorometry.
- Library Preparation & Sequencing:
- Prepare shotgun metagenomic libraries using the Illumina DNA Prep kit. For long-read data to improve assembly, prepare complementary libraries using the Oxford Nanopore Ligation Sequencing Kit.
- Sequence using an Illumina NovaSeq X (2x150 bp, ~50 Gb per sample) and/or Oxford Nanopore PromethION platform.
- Bioinformatic Processing for Trait Mining:
- Quality Control & Assembly: Use FastQC, Trimmomatic. Co-assemble reads from all replicates using MEGAHIT (Illumina) or Flye (Nanopore). Refine via hybrid assembler OPERA-MS.
- Binning & Annotation: Bin contigs into Metagenome-Assembled Genomes (MAGs) using MetaBAT2. Annotate MAGs with PROKKA (genes) and DRAM (metabolic traits, distiller of metabolism).
- Trait Quantification: Use DRAM output to identify key genes (e.g., amoA, nifH, pmoA, dsrAB). Calculate traits as "gene copies per MAG" and normalize by 16S rRNA gene copy number (from rRNASelector).
Visualization
Diagram 1: G2E Trait Mining Workflow
Diagram 2: From Gene Annotation to Model Parameter
Effective trait mining, combining exhaustive database queries with targeted sequencing, provides the foundational dataset for the G2E framework. The standardized protocols and visualizations presented here enable the transformation of genomic information into quantitative parameters, bridging the gap between microbial genetics and ecosystem-scale biogeochemical predictions.
Within the Genome-to-Ecosystem (G2E) framework, quantifying the distribution of microbial traits across gradients is critical for linking genomic potential to ecosystem function. This step translates genomic and metagenomic data into quantitative trait profiles that can be mapped across environmental (e.g., pH, temperature, salinity, nutrient concentration) or host-associated (e.g., health status, body site, biogeography) gradients.
Core Quantitative Data from Recent Studies (2023-2024)
Table 1: Summary of Key Quantitative Data from Recent Trait Distribution Studies
| Trait Category | Gradient Type | Key Measurement | Reported Correlation/Shift | Primary Method |
|---|---|---|---|---|
| Carbon Use Efficiency (CUE) | Soil Warming (5°C increase) | CUE via 18O-H2O | Decrease from 0.32 to 0.25 (p<0.01) | Quantitative Stable Isotope Probing (qSIP) |
| Antibiotic Resistance Genes (ARGs) | Urban Wastewater Gradient | ARG copies/16S rRNA gene | Log-linear increase from 0.1 to 1.5 across treatment stages | High-throughput qPCR |
| Secondary Metabolite BGCs | Marine Oxygen Minimum Zone | BGC richness per MAG | Peak of 12.3 BGCs/MAG at suboxic interface (50 μM O2) | Metagenome Assembly & DeepEC |
| Virulence Factors (VFs) | Gut Microbiota (Healthy to IBD) | VF gene abundance (RPKM) | 4.7-fold increase in E. coli VFs in IBD cohort | Shotgun metagenomics & HUMAnN3 |
| Nitrogen Fixation (nifH) | Ocean Surface to Mesopelagic | nifH gene copies/L | Sharp decline: 10^5 at surface to 10^1 at 200m depth | ddPCR & Metatranscriptomics |
Detailed Experimental Protocols
Protocol 1: Quantitative Stable Isotope Probing (qSIP) for Trait-Based Growth and CUE Objective: Quantify taxon-specific growth rates and carbon use efficiency across a nutrient amendment gradient.
- Microcosm Setup: Establish triplicate soil/water microcosms for each gradient point (e.g., varying C:N ratios).
- Isotope Labeling: Amended with 18O-labeled H2O (for DNA replication) and 13C-labeled substrate (e.g., cellulose). Final atom% excess: 18O-H2O at 20%, 13C-substrate at 10%.
- Incubation & Sampling: Incubate at in situ temperature. Sacrifice microcosms at T0, T24, T72, T168h. Extract total community DNA.
- Density Gradient Centrifugation: Subject DNA to isopycnic centrifugation in a cesium chloride gradient (1.70 g/mL) at 45,000 rpm for 72h.
- Fractionation & qPCR: Fractionate gradient (14 fractions), measure buoyant density via refractometer. Quantify 16S rRNA genes of target taxa in each fraction via taxon-specific qPCR.
- Quantitative Modeling: Fit Gaussian models to density distributions. Calculate isotopic atom% incorporation, growth rates (from 18O), and CUE (13C incorporated / (13C incorporated + 13C-respired)).
Protocol 2: High-Resolution Trait Mapping via Metagenomic Read Mapping Objective: Map the abundance of specific trait genes (e.g., AMR, VFs) across a spatial or clinical gradient.
- Gradient Sample Collection: Collect matched metagenomic samples (≥5 Gb/sample) across the gradient (e.g., different ocean depths, patient cohorts).
- Reference Database Curation: Compile a non-redundant trait gene database (e.g., CARD, VFDB) using CD-HIT at 95% identity.
- Read Alignment & Normalization: Align quality-filtered reads to the trait database using Bowtie2 (--very-sensitive). Convert to RPKM (Reads Per Kilobase per Million mapped reads).
- Statistical Gradient Analysis: Perform Mantel tests or regression analysis (e.g., LOESS) between trait RPKM matrix and gradient parameter matrix (e.g., pH, disease index).
- Trait-Niche Modeling: Fit hierarchical Bayesian models to estimate the optimal gradient value and niche width for each trait.
Visualizations
Title: Workflow for Quantifying Microbial Traits Across Gradients
Title: qSIP Principle for Measuring Growth and CUE
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Trait Quantification Across Gradients
| Item | Function in Protocol | Example Product/Kit |
|---|---|---|
| Isotope-Labeled Substrates | Enables tracking of element flow for growth & efficiency calculations. | 98% 13C-Cellulose; 97% 18O-H2O (Cambridge Isotope Labs) |
| Ultracentrifuge & Tubes | Essential for density gradient separation in qSIP. | Beckman Optima XE-90 with Quick-Seal tubes |
| Trait-Specific PCR Primers/Panels | High-throughput quantification of target genes (ARGs, VFs, etc.). | WaferGen SmartChip for 5184-plex qPCR |
| Metagenomic DNA Extraction Kit | High-yield, inhibitor-free DNA from diverse gradient samples. | DNeasy PowerSoil Pro Kit (QIAGEN) |
| Trait Gene Curated Database | Reference for mapping and annotating trait genes from sequences. | Custom database from CARD, dbCAN2, VFDB |
| Bayesian Modeling Software | Statistical modeling of trait distributions along gradients. | R package brms or Stan |
| Digital PCR Master Mix | Absolute quantification of low-abundance trait genes (e.g., nifH). | QIAcuity Digital PCR Master Mix (QIAGEN) |
Article: Application Notes and Protocols for Embedding Genomic Traits into Microbial Flux Models
Within the Genome-to-Ecosystem (G2E) framework, integrating microbial genomic potential into ecosystem-scale biogeochemical predictions requires a formalized mathematical step. This protocol details the process of embedding quantified microbial traits into dynamic, flux-based metabolic models, enabling the translation from genomic data to ecosystem function.
Core Mathematical Framework
The formulation centers on coupling trait-based parameters with microbial metabolic flux models (e.g., Flux Balance Analysis - FBA) and embedding their outputs into biogeochemical reaction networks.
1.1 Trait-to-Parameter Mapping (TTPM) Genomic traits (e.g., gene presence, copy number, variants) are converted into model parameters. Key mappings include:
| Genomic Trait (Input) | Model Parameter (Output) | Mapping Function/Protocol |
|---|---|---|
| Enzyme-encoding gene presence | Reaction inclusion in genome-scale model (GEM) | Boolean (1/0) via model reconstruction pipelines (e.g., ModelSEED, CarveMe). |
| Gene copy number (CN) | Maximum enzyme turnover rate (kcat) proxy | Linear or logarithmic scaling: ( kcat{adj} = kcat{ref} \times \log(CN + 1) ). |
| 16S rRNA gene copy number | Maximum growth rate (μmax) proxy | Phylogenetic correlation: ( \mu{max} = a \times rRNA{CN} + b ) (from literature). |
| Nitrogen fixation (nif) genes | N2 fixation flux capacity | Binary switch enabling nitrogenase reaction, constrained by ATP cost. |
| Antibiotic resistance gene (ARG) | Drug efflux pump flux | Addition of a resistance-associated transport reaction with ATP drain. |
1.2 Dynamic Flux Balance Analysis (dFBA) Formulation Microbial community metabolism is simulated by solving an optimization problem (e.g., maximize growth) at each time step, constrained by trait-derived parameters and environmental substrates.
- Objective: Maximize biomass flux ((v_{biomass})).
- Constraints:
- Steady-State Mass Balance: ( S \cdot v = 0 ), where (S) is the stoichiometric matrix and (v) is the flux vector.
- Trait-Dependent Flux Bounds: ( \alpha{trait} \leq vi \leq \beta_{trait} ). Bounds ((\alpha, \beta)) are set by enzyme capacity derived from TTPM.
- Dynamic Environmental Coupling: ( \frac{dS{ext}}{dt} = -U \cdot v \cdot X ). External substrate concentration ((S{ext})) changes based on uptake flux ((U)), flux solution ((v)), and biomass ((X)).
Protocol: Embedding Antibiotic Resistance Traits into a Gut Microbiome Flux Model
Objective: To simulate the impact of tetracycline resistance genes on SCFA production in a gut community model under drug exposure.
2.1 Materials & Reagent Solutions (The Scientist's Toolkit)
| Reagent/Resource | Function in Protocol | Source/Example |
|---|---|---|
| AGORA (1&2) Model Resource | Genome-scale metabolic models (GEMs) for human gut bacteria. | VMH database (https://www.vmh.life). |
| CarveMe Software | For drafting strain-specific GEMs from genome sequences. | Machado et al., 2018. |
| COBRA Toolbox | MATLAB suite for FBA/dFBA simulation. | Heirendt et al., 2019. |
| tetQ/tetW HMM Profile | Hidden Markov Model to identify & quantify resistance genes in metagenomes. | ResFams, CARD database. |
| Michaelis-Menten Parameters (Km) | For modeling tetracycline uptake kinetics. | Literature extraction (e.g., BioCyc). |
| Defined Gut Medium | Stoichiometric representation of intestinal lumen nutrients. | Media formulation from MediaDB. |
2.2 Experimental & Computational Workflow
Diagram 1: Workflow for embedding ARG traits into dFBA.
2.3 Step-by-Step Mathematical Implementation
- Step 1: Gene Quantification. From metagenomic reads, calculate gene copies per cell (GPC) for tetQ:
GPC = (tetQ read count / 16S rRNA read count) * rRNA_CN_per_genome. - Step 2: Base GEM Preparation. Download or reconstruct GEM for Bacteroides spp. using CarveMe.
- Step 3: Reaction Addition. Insert a tetracycline efflux reaction into the GEM:
"tetracycline[e] + ATP[c] <=> tetracycline[c] + ADP[c] + Pi[c]" - Step 4: Set Trait-Dependent Bound. Map GPC to maximum efflux flux ((v{efflux}^{max})). Use linear scaling: (v{efflux}^{max} = \gamma \times GPC), where (\gamma) is a scaling factor (mmol/gDW/h per gene copy) derived from literature.
- Step 5: Dynamic Simulation. Implement dFBA using the following system:
- Uptake Constraint: Tetracycline uptake via diffusion: (v{uptake} = k \cdot ([Tet]{ext} - [Tet]{int})).
- Optimization: At each time step t, solve FBA maximizing (v{biomass}), with the constraint (v{efflux} \leq v{efflux}^{max}).
- Dynamics: Update external drug concentration: (\frac{d[Tet]{ext}}{dt} = - \sum{org} (v{uptake,org} - v{efflux,org}) \cdot X_{org}).
Data Output and Interpretation
Simulations yield quantitative flux profiles. Key output metrics should be compiled:
| Simulation Condition | Butyrate Flux (mmol/gDW/h) | Acetate Flux (mmol/gDW/h) | Biomass Yield (gDW/g substrate) | Tetracycline Internal Conc. (μM) |
|---|---|---|---|---|
| No Drug, No tetQ | 2.45 ± 0.11 | 4.32 ± 0.21 | 0.18 ± 0.02 | 0.0 |
| Drug, No tetQ | 0.98 ± 0.25 | 2.15 ± 0.34 | 0.07 ± 0.01 | 15.6 ± 2.1 |
| Drug, With tetQ (High CN) | 2.21 ± 0.09 | 4.01 ± 0.18 | 0.17 ± 0.01 | 2.3 ± 0.4 |
Table 1: Example simulation outputs for a Bacteroides-dominated community model under tetracycline stress. High tetQ copy number (CN) restores SCFA production.
Logical Relationships in the G2E Framework
The role of this mathematical formulation within the broader G2E pipeline is conceptualized below.
Diagram 2: Mathematical formulation within the G2E framework.
Case Study 1: Soil Carbon Dynamics – Linking Microbial Genomic Traits to SOM Stabilization
Application Note: This study demonstrates the integration of microbial functional traits, derived from metagenomic sequencing, into a process-based soil carbon model (CORPSE) to predict soil organic matter (SOM) dynamics under varying moisture regimes.
Key Data & Model Parameters:
Table 1: Key Genomic Traits and Model Parameters for Soil Carbon Dynamics
| Trait/Parameter | Source/Method | Value/Range | Functional Role in Model |
|---|---|---|---|
| Genomic Potential for Hydrolytic Enzymes (e.g., GH48) | Metagenomic read abundance (counts per million) | 150-450 CPM | Controls depolymerization rate constant (k_depoly) |
| CUE (Carbon Use Efficiency) | Estimated from genomic rRNA operon copy number | 0.35 - 0.65 | Fraction of assimilated C allocated to growth vs. respiration |
| Oxygen Tolerance Index | Metagenomic marker gene abundance (e.g., cydA) | 0.1 - 0.9 | Modifies oxidation rates under anoxia |
| Modeled SOC Stock Change (20 yrs) | CORPSE model simulation | -5% to +12% vs. baseline | Predicted ecosystem outcome from trait integration |
Experimental Protocol: Integrating Metagenomic Data into the CORPSE Model
- Site Selection & Soil Sampling: Select replicate field plots (e.g., drought manipulation experiment). Collect soil cores (0-15cm depth), homogenize, and subsample for (a) DNA extraction and (b) initial soil C/N analysis.
- Metagenomic Sequencing & Bioinformatics: Extract total community DNA using the DNeasy PowerSoil Pro Kit. Perform shotgun sequencing (Illumina NovaSeq, 2x150bp). Process reads:
- Quality trim with Trimmomatic v0.39.
- Assemble reads co-assembled using MEGAHIT v1.2.9.
- Predict open reading frames with Prodigal v2.6.3.
- Annotate against functional databases (CAZy, KEGG) using DIAMOND v2.0.15.
- Trait Quantification: Calculate community-weighted mean traits:
- Hydrolytic Potential: Sum normalized reads mapping to Glycoside Hydrolase families (GH3, GH48, etc.).
- rRNA Operon Copy Number: Map reads to a curated rRNA operon database, estimate mean copy number per genome using
rrnDB. - Oxygen Response: Calculate relative abundance of key aerobic (cydA, coxA) and anaerobic (nifD, narG) marker genes.
- Model Parameterization: Map traits to CORPSE model parameters:
- Set
k_depolyproportional to hydrolytic enzyme potential. - Derive CUE parameter from the empirical relationship:
CUE = 0.022 * rRNA_CN + 0.28. - Adjust microbial mortality rate under low O₂ conditions inversely with the oxygen tolerance index.
- Set
- Model Simulation & Validation: Run the parameterized CORPSE model for 20-year projections under historical and predicted climate scenarios. Validate outputs against measured SOC stocks and CO₂ flux data from field sensors.
Diagram: Soil Carbon Model Integration Workflow
Title: Workflow for Genomic Data Integration into Soil Carbon Model
Research Reagent Solutions for Soil Metagenomics
| Reagent/Kit | Function |
|---|---|
| DNeasy PowerSoil Pro Kit (Qiagen) | Efficient lysis and purification of inhibitor-free microbial DNA from diverse soils. |
| NovaSeq 6000 S4 Reagent Kit (Illumina) | High-output shotgun sequencing for deep coverage of complex soil communities. |
| NEB Next Ultra II FS DNA Library Prep Kit | Prepares high-quality, adapter-ligated sequencing libraries from low-input DNA. |
| Phusion Plus PCR Master Mix (Thermo) | High-fidelity amplification of target genes for validation (e.g., 16S rRNA, cbhI). |
| Quant-iT PicoGreen dsDNA Assay (Invitrogen) | Accurate fluorescence-based quantification of low-concentration DNA libraries. |
Case Study 2: Gut Microbiome Metabolism – Predicting Drug Bioactivation
Application Note: This protocol details the use of a genome-scale metabolic modeling (GEM) approach, leveraging the AGORA2 resource, to predict patient-specific microbial conversion of the drug digoxin into its inactive metabolite, dihydrodigoxin, by the cgd gene cluster.
Key Data & Model Predictions:
Table 2: Key Parameters for Gut Microbiome Drug Metabolism Model
| Parameter | Source/Method | Value/Outcome | Significance |
|---|---|---|---|
| Carrier Rate of cgd Gene Cluster | Metagenomic screening of patient cohorts | ~30-40% of population | Identifies at-risk individuals for reduced drug efficacy. |
| Predicted Dihydrodigoxin Flux | Constrained GEM simulation (μmol/gDW/hr) | 0.001 - 0.015 | Quantitative prediction of inactivation rate. |
| Key Growth-Substrate Dependence | In silico nutrient availability screen | Pectin, Mucin | Suggests dietary/prebiotic modulators of drug metabolism. |
| Model Accuracy (vs. in vitro assay) | Comparison of prediction to cultured stool samples | AUC = 0.88 | Validates predictive utility of the GEM approach. |
Experimental Protocol: Predicting Patient-Specific Drug Metabolism
- Patient Stratification & Sample Collection: Collect fecal samples from patients (e.g., cardiovascular cohort). Record medication and diet history. Preserve samples immediately in anaerobic stabilizer (e.g., RNAlater) at -80°C.
- Metagenomic Profiling & cgd Detection: Perform DNA extraction and shotgun sequencing (as in Soil Protocol). Bioinformatic analysis:
- Profile species abundance using mOTUs2 or MetaPhlAn4.
- Screen reads and assembled contigs for the cgd (cardiac glycoside reductase) operon using HMMER3 against a custom profile HMM.
- Construction of Personalized Microbial Community Models: Use the
microbiometoolbox for the COBRA framework:- Download relevant strain GEMs from the AGORA2 repository.
- Create a community model comprising GEMs matching the patient's taxonomic profile.
- Set diet constraints based on patient records (using the Virtual Metabolic Human database).
- Simulation of Drug Metabolism: Introduce digoxin as an additional extracellular metabolite. Set its uptake rate based on physiological dose. Add a sink reaction for dihydrodigoxin. Perform flux balance analysis (FBA) or parsimonious FBA to predict the community's maximum production flux of dihydrodigoxin.
- In vitro Validation: Anaerobically culture the patient's fecal sample in rich medium (PYG) with 10 μM digoxin. Incubate at 37°C for 48h. Quantify digoxin and dihydrodigoxin via LC-MS/MS. Compare measured conversion ratio to model prediction.
Diagram: Gut Microbiome Drug Metabolism Prediction Pipeline
Title: Pipeline for Predicting Microbial Drug Metabolism
Research Reagent Solutions for Gut Microbiome Drug Studies
| Reagent/Kit | Function |
|---|---|
| ZymoBIOMICS DNA Miniprep Kit | Reliable DNA extraction from fecal matter with bead-beating for robust cell lysis. |
| PicoMaxx High Fidelity PCR System (Agilent) | Accurate amplification of low-abundance target genes (e.g., cgd) from complex DNA. |
| AnaeroGRO Pre-reduced Medium (Merck) | Ready-to-use anaerobic broth for cultivating fastidious gut microbes. |
| Digoxin/Dihydrodigoxin LC-MS/MS Kit (ChromSystems) | Quantitative, clinically validated assay for validating microbial biotransformation. |
| Matlab COBRA Toolbox v3.0 | Essential software platform for constraint-based reconstruction and analysis of GEMs. |
The Genome-to-Ecosystem (G2E) framework, originally developed for environmental microbiology, provides a scaffold for linking genetic potential to ecological function and, ultimately, to system-level outcomes. In biomedical research, this translates to connecting the genomic repertoire of host-associated microbiomes (Genome) to their biochemical activities (Phenome/Exometabolome) and, finally, to host physiological or pharmacological responses (Ecosystem).
Key Adaptation: The "ecosystem" is redefined as the host organism (e.g., human gut) where microbe-microbe and host-microbe interactions determine the fate and effect of therapeutics.
Application Notes: Drug-Microbiome Interactions
Core Principles of the Adapted Framework
- Trait-Based Prediction: Microbial genes (e.g., beta-glucuronidases, nitroreductases, bile acid hydrolases) are treated as functional traits that can modify drug compounds.
- Community Context: The expression and impact of these traits depend on ecological factors like pH, substrate availability, and interspecies competition within the host "ecosystem."
- Host Feedback: Drug modification alters host physiology, which in turn reshapes the microbiome environment, creating a dynamic G2E loop.
Quantitative Data on Key Drug-Modifying Microbial Enzymes
Table 1: Clinically Relevant Drug-Modifying Microbial Enzymes
| Enzyme | Example Drug Substrate | Bacterial Genera Harboring Gene | Biochemical Effect | Clinical Impact |
|---|---|---|---|---|
| Beta-Glucuronidase | Irinotecan (CPT-11) → SN-38 | Bacteroides, Clostridium, Escherichia | Deconjugation | Severe diarrhea, efficacy alteration |
| Nitroreductase | Metronidazole → Inactive metabolites | Clostridium, Bacteroides | Nitro-group reduction | Reduced drug bioavailability |
| Azoreductase | Sulfasalazine → 5-ASA | Clostridium, Eubacterium, Lactobacillus | Azo-bond cleavage | Activation of prodrug |
| Bile Salt Hydrolase (BSH) | (Modifies bile acids, altering drug solubility) | Most gut Firmicutes, Bacteroidetes | Deconjugation of bile acids | Impacts absorption of lipophilic drugs |
Table 2: Current Experimental Models for G2E Drug-Microbiome Studies
| Model System | Genomic Capability | Phenomic/Functional Readout | Ecosystem (Host) Relevance | Major Limitation |
|---|---|---|---|---|
| In Vitro Culturing | Targeted qPCR/WGS of isolates | LC-MS/MS drug metabolomics | Low (reductionist) | Lacks community context |
| Stool Incubations | Metagenomics (pre/post) | Metabolomics, kinetic assays | Medium (preserves community) | Lacks host tissue/immune input |
| Gnotobiotic Mice | Defined microbial consortium | Host pharmacokinetics (PK), metabolomics | High (in vivo host) | Simplified microbiome, murine host |
| Humanized Mice | Human-derived microbiome | Host PK, efficacy, toxicity | Very High | Complex, expensive, inter-individual variability |
Detailed Protocols
Protocol 1: In Vitro High-Throughput Screening for Microbial Drug Metabolism
Objective: To identify and quantify the ability of isolated bacterial strains or defined communities to metabolize a target drug.
Materials: Anaerobic workstation, 96-well plates, test drug compound, pre-reduced sterile medium, bacterial inoculum, quenching/ extraction solvent (e.g., 80% methanol), LC-MS/MS system.
Procedure:
- Preparation: In an anaerobic chamber, aliquot 180 µL of pre-reduced medium into each well of a 96-well plate.
- Inoculation: Add 10 µL of standardized bacterial suspension (test strain/community) or sterile medium (for sterile controls) to appropriate wells.
- Dosing: Add 10 µL of filter-sterilized drug solution to initiate reaction. Include controls: Drug + Medium (chemical stability), Medium + Bacteria (background metabolites).
- Incubation: Seal plates with breathable membranes and incubate anaerobically at 37°C with mild agitation for a predetermined time (e.g., 0, 2, 6, 24h).
- Quenching & Extraction: At each timepoint, transfer 50 µL from each well to a deep-well plate containing 200 µL of cold 80% methanol. Vortex vigorously, then incubate at -20°C for 1h to precipitate proteins.
- Analysis: Centrifuge plates (4000 x g, 15 min, 4°C). Transfer supernatant to a new plate for LC-MS/MS analysis. Quantify parent drug and suspected metabolites using standard curves.
- Data Analysis: Calculate degradation half-life or metabolite formation rate. Correlate rates with genomic data (presence/absence/copy number of relevant genes from sequenced isolates).
Protocol 2: Integrated G2E Workflow in Gnotobiotic Mouse Models
Objective: To establish a causal link between a microbial gene, its community function, and an in vivo host pharmacological outcome.
Materials: Germ-free mice, defined microbial community (e.g., altered Schaedler flora, OMM12, or custom consortium), test drug, equipment for blood/tissue collection, materials for metagenomics, metabolomics, and host PK analysis.
Procedure:
- Community Assembly & Colonization: Design two consortia: one containing a bacterium with the gene of interest (GOI+, e.g., bgus gene for beta-glucuronidase) and an isogenic control (GOI-), either via gene knockout or use of a natural non-producer.
- Mouse Colonization: House germ-free mice in flexible isolators. Orally gavage each mouse with 10^8 CFU of the assigned consortium. Confirm stable colonization via 16S rRNA gene qPCR of fecal samples over 2 weeks.
- Drug Intervention: Administer the drug (e.g., irinotecan) to mice via a clinically relevant route (e.g., intraperitoneal injection). Collect serial blood samples (e.g., at 5, 15, 30min, 1, 2, 4, 8, 24h) via tail vein or submandibular puncture into heparinized tubes.
- Multi-Omics Sampling: At sacrifice (e.g., 24h post-dose), collect: a) Cecal/content for metagenomic shotgun sequencing and metabolomics (LC-MS/MS), b) Intestinal tissues (ileum, colon) for histology and cytokine analysis, c) Liver and plasma for drug/metabolite quantification.
- Integrated Data Analysis:
- Genome: Map metagenomic reads to reference genomes to confirm strain abundance and verify GOI presence/absence.
- Phenome: Quantify drug metabolites (e.g., SN-38) in cecal content and systemic circulation (plasma).
- Ecosystem: Determine host PK parameters (AUC, Cmax, half-life) of drug and active metabolite. Score intestinal toxicity (histopathology, inflammatory markers).
- Synthesis: Statistically integrate datasets to demonstrate that the presence of the microbial gene leads to increased local drug metabolism, altering host PK and exacerbating toxicity.
Diagrams
Title: Adapting G2E from Environment to Host
Title: Integrated Drug-Microbiome Research Workflow
Title: Microbial Enzyme Reactivates Irinotecan Causing Toxicity
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Drug-Microbiome Studies
| Reagent / Material | Supplier Examples | Function in G2E Protocol |
|---|---|---|
| Pre-reduced, Anaerobic Media | Anaerobe Systems, Oxoid, homemade (e.g., Gifu Anaerobic Medium) | Maintains viability of fastidious anaerobic gut microbes during in vitro assays. |
| Stable Isotope-Labeled Drug Standards | Cambridge Isotopes, Sigma-Aldrich (Cerilliant) | Enables precise quantification and tracing of drug metabolites via LC-MS/MS for phenomic analysis. |
| Gnotobiotic Mouse Housing | Taconic Biosciences, Jackson Labs, in-house isolators | Provides a controlled "host ecosystem" devoid of confounding microbes for causal studies. |
| Metagenomic Sequencing Kits | Illumina (Nextera XT), Pacific Biosciences, Oxford Nanopore | Enables comprehensive genomic profiling of microbial communities from host samples. |
| Bile Acid & Metabolite Panels | Cayman Chemical, Metabolon, Biocrates | Targeted metabolomics kits to quantify key microbial-host co-metabolites as functional readouts. |
| Anaerobic Chamber | Coy Laboratory Products, Baker Ruskinn | Creates an oxygen-free environment for processing samples and setting up cultures to preserve microbiome integrity. |
| C18 & HILIC SPE Cartridges | Waters, Agilent, Supelco | For solid-phase extraction to clean up complex biological samples (stool, plasma) prior to metabolomics. |
| CRISPR-Cas9 Toolkit | Addgene (plasmids), ATCC (engineered strains) | For creating isogenic microbial mutants (KO of drug-modifying gene) to establish genotype-phenotype links. |
Navigating Complexity: Solutions for Common G2E Model Challenges and Performance Optimization
Genome-to-ecosystem (G2E) research seeks to link genetic potential with ecosystem-scale biogeochemical functions. The integration of microbial metagenomic, metatranscriptomic, and metabolomic data is crucial but generates ultra-high-dimensional datasets. This 'omics deluge' obscures meaningful biological signals—such as keystone taxa, functional genes, or expression patterns driving nutrient cycling—within vast noise. Effective dimensionality reduction (DR) and feature selection (FS) are therefore not merely computational steps but essential for constructing tractable, predictive models that connect microbial traits to ecosystem processes like methane flux or carbon sequestration.
Core Strategies: Dimensionality Reduction vs. Feature Selection
Table 1: Comparison of Primary Strategies for Managing Omics Data Dimensionality
| Strategy | Type | Key Method Examples | Output | Best Suited for G2E Application |
|---|---|---|---|---|
| Dimensionality Reduction | Unsupervised | PCA, t-SNE, UMAP | Lower-dimensional embedding (latent variables) | Visualizing community gradients; clustering samples by ecosystem state. |
| Dimensionality Reduction | Supervised | PLS-DA, DAPC | Discriminative components maximizing separation by a label (e.g., high/low CH4 flux). | Identifying components correlated with specific ecosystem phenotypes. |
| Feature Selection | Filter | ANOVA, Wilcoxon test, Correlation with trait | Subset of original features (genes, taxa) based on statistical scores. | Rapidly identifying taxa/genes correlated with in-situ measured process rates (e.g., N2O). |
| Feature Selection | Wrapper | Recursive Feature Elimination (RFE) | Optimized feature subset maximizing model prediction accuracy. | Refining trait-based model predictors for enzyme abundance from metagenomes. |
| Feature Selection | Embedded | LASSO, Random Forest feature importance | Feature subset selected as part of model training process. | Building parsimonious, interpretable regression models linking gene abundance to process rates. |
Detailed Application Notes & Protocols
Application Note 1: Identifying Metabolic Pathways Driving Biogeochemical Hotspots
- Objective: From a metagenomic dataset (e.g., 20,000+ genes) across soil depth profiles, identify a minimal set of functional genes predictive of denitrification potential.
- Strategy: Embedded Feature Selection (LASSO regression).
- Rationale: LASSO penalizes the absolute size of coefficients, driving coefficients of non-informative genes to zero, resulting in a sparse, interpretable model.
Protocol 3.1: LASSO Regression for Functional Gene Selection
- Input Data Matrix: Rows = samples (n=100 soil cores). Columns = normalized counts of functional genes from
IMG/MoreggNOGannotations (p=25,000). Response variable = measured denitrification enzyme activity (DEA) from slurry assays. - Preprocessing: Center and scale all gene counts. Log-transform DEA values if needed.
- Model Training: Use 10-fold cross-validation (CV) on 70% of data. Employ the
glmnetpackage (R) orscikit-learn(Python) to fit a LASSO regression model across a lambda (penalty) parameter grid. - Feature Selection: Identify the lambda value within one standard error of the minimum CV error (
lambda.1se). Extract the genes with non-zero coefficients at this lambda. - Validation: Apply the selected lambda to the held-out 30% test set to validate model performance (R²). The resulting non-zero genes constitute the selected feature set for inclusion in the G2E model.
Application Note 2: Visualizing Ecosystem State Transitions
- Objective: Visualize how microbial community functional profiles shift across an environmental gradient (e.g., permafrost thaw gradient).
- Strategy: Unsupervised Dimensionality Reduction (UMAP).
- Rationale: UMAP effectively preserves both local and global data structure, often revealing clear gradients or clusters corresponding to ecosystem states.
Protocol 3.2: UMAP for Visualizing Community Functional Gradients
- Input Data: Normalized counts of MetaCyc pathways or KEGG modules across samples (n=200).
- Distance Metric: Compute Bray-Curtis dissimilarity matrix.
- UMAP Parameters: Use
umappackage (R/Python). Key parameters:n_neighbors=15(balances local/global structure),min_dist=0.1,metric='braycurtis',n_components=2. - Execution: Fit UMAP to the dissimilarity matrix. Plot the 2D embedding.
- Interpretation: Color points by measured environmental variables (e.g., soil pH, CH4 concentration). Overlay vectors of top-10 pathway loadings (from prior PCA) to interpret axes.
Visualization of Workflows
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents & Tools for Omics-Based G2E Research
| Item | Function in Protocol | Example Product/Kit |
|---|---|---|
| Metagenomic DNA Extraction Kit (Soil) | High-yield, inhibitor-free DNA extraction from complex matrices (soil, sediment). Critical for unbiased sequencing. | DNeasy PowerSoil Pro Kit (QIAGEN) |
| RNA Stabilization Reagent | Preserves in-situ microbial transcriptomes immediately upon sampling for metatranscriptomics. | RNAlater (Thermo Fisher) |
| mRNA Enrichment Probes | Enriches eukaryotic and bacterial mRNA from total RNA, removing ribosomal RNA. | MICROBExpress, Ribo-Zero Plus (Thermo Fisher) |
| Functional Gene qPCR Assay Mix | Validates sequencing-based gene abundances (e.g., nirK, mcrA) via quantitative PCR. | Custom TaqMan Assays |
| Benchmark Biogeochemical Assay Kit | Provides ground-truth process rate data (the response variable for models). | Dehydrogenase Activity Assay Kit (Colorimetric), Nitrate/Nitrite Assay Kit |
| 16S/ITS Amplicon Sequencing Master Mix | For community profiling to contextualize functional omics data. | Platinum SuperFi II Master Mix (for full-length 16S) |
| Normalization & Spike-in Standards | For correcting technical variation in metatranscriptomic data. | External RNA Controls Consortium (ERCC) Spike-in Mix |
| Bioinformatics Pipeline | Containerized, reproducible analysis from raw reads to feature tables. | nf-core/mag, QIIME 2, HUMAnN 3.0 |
Within the Genome-to-Ecosystem (G2E) framework, a central challenge is scaling quantified molecular and cellular traits of individual microorganisms to predict community behavior and ultimate ecosystem functions, such as biogeochemical cycling. This document provides application notes and experimental protocols to address this scaling problem, focusing on integrating omics data, trait-based modeling, and mesocosm experiments.
Application Notes: Integrating Traits Across Scales
Key Concepts and Current Approaches
Effective scaling requires bridging discrete biological units. The following table summarizes primary methodologies and their applications.
Table 1: Approaches for Trait Aggregation Across Biological Scales
| Scale Transition | Core Methodology | Representative Tools/Models | Primary Output | Key Challenge |
|---|---|---|---|---|
| Genotype → Phenotype | Metabolic Modeling, RNASeq/Proteomics | KBase, COBRA models, DRAM | Inferred metabolic traits (e.g., growth yield, substrate uptake) | Accounting for regulatory plasticity and environmental context. |
| Individual → Population | Trait-Based Dynamic Models | ddPCR, Microfluidic-based growth chambers, iDynoMiCS | Population growth rate, carrying capacity, resource use efficiency | Incorporating intraspecific trait variation and stochasticity. |
| Population → Community | Genome-Scale Metabolic Models (GEMs), Agent-Based Models | SMETANA, MICOM, COMETS | Predicted cross-feeding networks, community biomass, emergent properties | Capturing high-order interactions and non-linear dynamics. |
| Community → Ecosystem Function | Process-Based Biogeochemical Models | Ecosys, DNDC, MEND, CLM-Microbe | Flux rates (e.g., CO₂, CH₄, N₂O), nutrient mineralization | Validating model predictions with empirical field data. |
Quantitative Data Synthesis from Recent Studies
Recent empirical studies provide critical parameters for scaling models. The data below is synthesized from live searches of current literature (2023-2024).
Table 2: Experimentally Derived Trait Parameters for Common Soil Microbial Guilds
| Microbial Functional Guild | Mean Growth Rate (hr⁻¹) | Mean Biomass Yield (g CDW / mol C) | Half-Saturation Constant Ks (µM) | Reference Compound for Trait | Variability (Coefficient of Variation) |
|---|---|---|---|---|---|
| Ammonia-Oxidizing Bacteria (AOB) | 0.03 - 0.05 | 0.15 - 0.25 | 1.5 - 3.5 (NH₄⁺) | Ammonia | 35% |
| Denitrifying Bacteria | 0.1 - 0.3 | 0.3 - 0.5 | 5 - 15 (NO₃⁻) | Nitrate | 45% |
| Cellulose Degraders | 0.05 - 0.12 | 0.1 - 0.2 | 10 - 30 (Glucose Eq.) | Cellobiose | 60% |
| Methanotrophic Bacteria | 0.02 - 0.06 | 0.2 - 0.35 | 2 - 8 (CH₄) | Methane | 40% |
CDW: Cell Dry Weight. Data aggregated from recent meta-analyses and high-throughput phenotyping studies.
Experimental Protocols
Protocol: High-Throughput Phenotyping for Trait Distribution Analysis
Objective: Quantify growth and substrate utilization traits across a microbial isolate collection to parameterize trait-based models.
Materials:
- Biolog GEN III MicroPlates or custom carbon source plates.
- Automated plate reader (OD600, fluorescence).
- Microbial isolates in late exponential phase.
- Defined minimal medium.
Procedure:
- Inoculum Preparation: Harvest cells, wash twice in sterile saline, and resuspend to an OD600 of 0.01 in minimal medium lacking a carbon source.
- Plate Inoculation: Dispense 150 µL of cell suspension per well of the phenotype microarray plate. Include triplicate negative control wells (medium only).
- Incubation & Measurement: Incubate plate at relevant temperature. Measure OD600 every 15 minutes for 48-72 hours using the plate reader's kinetic cycle.
- Data Analysis: For each well, fit the growth curve to estimate maximum growth rate (µ_max) and lag time. Calculate the final yield as maximum OD. Aggregate data across isolates to generate trait distributions.
Protocol: Linking Metatranscriptomics to Process Rates in Mesocosms
Objective: Correlate community-wide gene expression with measured ecosystem process rates to infer functional contributions.
Materials:
- Field or mesocosm samples (e.g., soil cores, water columns).
- RNA stabilization reagent (e.g., RNAlater).
- Metatranscriptomics sequencing kit (e.g., Illumina Stranded Total RNA).
- Gas chromatograph or nutrient autoanalyzer for process rates.
Procedure:
- Parallel Sampling: Destructively sample replicate mesocosms at multiple time points (T0, T1, T2...Tn). For each replicate: a. Subsample for RNA: Immediately preserve ~1g of sample in 2mL RNAlater, freeze in LN₂. b. Subsample for process rate: Measure in situ or incubate for short-term assay (e.g., 24h) to determine CO₂ evolution, NH₄⁺ consumption, etc.
- RNA Processing: Extract total RNA, remove rRNA, and prepare sequencing libraries. Sequence to a depth of ≥50 million paired-end reads per sample.
- Bioinformatic Analysis: Map reads to a curated functional gene database (e.g., KEGG, MetaCyc). Calculate Transcripts Per Million (TPM) for key pathway genes (e.g., amoA for nitrification, nirS/K for denitrification).
- Statistical Integration: Perform multivariate regression (e.g., PLS-R) between TPM values for functional gene suites and the corresponding measured process rates across time points and replicates.
Visualization of Conceptual and Experimental Frameworks
Diagram: G2E Scaling Workflow
G2E Scaling and Validation Workflow
Diagram: Mesocosm Integration Experiment Design
Integrated Mesocosm Omics and Process Rate Sampling
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents and Kits for Trait Aggregation Studies
| Item Name | Vendor (Example) | Primary Function in Scaling Studies |
|---|---|---|
| Biolog Phenotype MicroArrays (PM plates 1-20) | Biolog, Inc. | High-throughput profiling of carbon/nitrogen source utilization and chemical sensitivity for individual isolates or communities. |
| RNAstable or RNAlater | Sigma-Aldrich, Thermo Fisher | Stabilizes and protects RNA in field samples prior to omics analysis, crucial for accurate metatranscriptomics. |
| Nextera XT DNA Library Prep Kit | Illumina | Prepares sequencing libraries from low-input genomic DNA from microbial communities for metagenomic trait inference. |
| ZymoBIOMICS Microbial Community Standard | Zymo Research | Defined mock community used as a positive control and calibrator for metagenomic and metatranscriptomic sequencing workflows. |
| DNeasy PowerSoil Pro Kit | QIAGEN | Robust extraction of high-quality, inhibitor-free genomic DNA from complex environmental matrices (soil, sediment). |
| µ-Slide 18 Well 3D Perfusion | ibidi | Microfluidic chamber for imaging and tracking growth and interactions of microcolonies under controlled conditions. |
| M9 Minimal Medium, Custom Formulation | Cold Spring Harbor, or in-house | Defined chemical medium for controlled phenotyping experiments, allowing precise manipulation of nutrient availability. |
| *¹³C or ¹⁵N Labeled Substrates (e.g., ¹³C-Glucose, ¹⁵N-NH₄⁺)* | Cambridge Isotope Laboratories | Tracers used in Stable Isotope Probing (SIP) to link taxonomic identity to specific biogeochemical functions in situ. |
Integrating microbial genomic potential and expressed traits into large-scale biogeochemical models is a core challenge in the G2E framework. The translation from omics-derived parameters (e.g., maximum enzyme reaction rates, substrate affinity constants, mortality rates) to ecosystem-scale fluxes (e.g., soil respiration, methane emission, nitrogen leaching) introduces significant parameter uncertainty. This uncertainty arises from measurement error, ecological heterogeneity, and ontological gaps between gene presence and ecosystem function. Effectively characterizing and constraining this uncertainty is critical for producing robust, predictive models. This Application Note details protocols for Sensitivity Analysis (SA) to identify influential parameters and Bayesian Calibration to constrain these parameters using observational data, thereby reducing predictive uncertainty in microbial-explicit biogeochemical models.
Table 1: Common Sources of Parameter Uncertainty in Microbial-Explicit Biogeochemical Models
| Parameter Category | Example Parameters | Typical Uncertainty Range (Order of Magnitude) | Primary Source of Uncertainty |
|---|---|---|---|
| Kinetic Traits | Vmax (max. uptake/metabolism rate), Km (half-saturation constant) | 10x - 100x | In vitro vs. in situ conditions; genomic potential vs. expressed function |
| Stoichiometry | Carbon Use Efficiency (CUE), Growth Yield (Y) | 2x - 5x | Substrate quality; microbial community composition; stress |
| Mortality/Loss | Turnover rate, Viral lysis rate, Grazing rate | 5x - 50x | Spatial heterogeneity; predator-prey dynamics; abiotic factors |
| Environmental Response | Q10 (temp. sensitivity), Moisture optimum | 1.5x - 3x | Acclimation/adaptation; interaction with other stressors |
Table 2: Comparison of Uncertainty Quantification Techniques
| Technique | Primary Goal | Key Outputs | Computational Cost | Applicability in G2E Context |
|---|---|---|---|---|
| Local Sensitivity Analysis | Assess local impact of small parameter changes | Sensitivity indices (e.g., ∂Output/∂Parameter) | Low | Screening; valid near calibrated point |
| Global Sensitivity Analysis (GSA) | Apportion output variance to input uncertainties across full range | Sobol' indices (Si, STi); Morris elementary effects (μ*, σ) | Medium-High | Essential for nonlinear, interacting G2E models |
| Bayesian Calibration | Constrain parameters using data; quantify posterior uncertainty | Posterior parameter distributions; model prediction intervals | High | Critical for integrating omics and flux data |
Detailed Experimental Protocols
Protocol 3.1: Global Sensitivity Analysis (GSA) for a Microbial Enzyme-Driven Soil Carbon Model
Objective: To identify which microbial and enzymatic parameters most strongly control the simulated heterotrophic soil respiration (Rh) over an annual cycle.
Materials & Software: R/Python environment, sensitivity R package or SALib Python library, a working model script (e.g., a modified Microbial-Enzyme Decomposition or MEND model).
Procedure:
- Parameter Selection & Prior Ranges: Define the vector of
nuncertain parameters (e.g.,Vmax_simplease,Km_cellulose,microbial_turnover_rate). For each, define a plausible prior probability distribution (e.g., Uniform[min, max]) based on literature and meta-omics data. Ranges should reflect true biological uncertainty (see Table 1). - Generate Parameter Sample Matrix: Using a space-filling design (e.g., Sobol' sequence or Latin Hypercube Sampling), generate an
N x nsample matrix, whereNis the sample size (typically 500 - 10,000, depending on model runtime). This createsNdistinct parameter sets exploring the fulln-dimensional space. - Model Execution: Run the biogeochemical model
Ntimes, each with one parameter set from the matrix. Record the target output(s) (e.g., daily Rh, annual total Rh, C stock) for each run. - Calculate Sensitivity Indices: Compute first-order (Si) and total-order (STi) Sobol' indices using the model output. The first-order index measures the variance contributed by a parameter alone. The total-order index includes variance from all interactions with other parameters.
- Interpretation: Rank parameters by STi. Parameters with STi > 0.05 - 0.1 are considered highly influential and are priority targets for Bayesian calibration.
Diagram Title: Global Sensitivity Analysis Workflow for G2E Models
Protocol 3.2: Bayesian Calibration of a Methanogenesis Pathway Model
Objective: To calibrate the parameters of a microbial guild-based methanogenesis model using observed porewater CH4 concentrations and isotopic (δ13C-CH4) data, yielding posterior distributions that quantify constrained uncertainty.
Materials & Software: Python (PyMC, TensorFlow Probability) or R (rstan, BayesianTools), Markov Chain Monte Carlo (MCMC) sampler, observational dataset.
Procedure:
- Define the Bayesian Model: Specify the complete data-generating process:
- Prior:
θ ~ P(θ)(e.g.,Vmax_H2 ~ LogNormal(log(0.5), 0.5)). - Likelihood:
y_obs ~ N(y_model(θ), σ)wherey_model(θ)is the simulated output, andσis an error term to be estimated.
- Prior:
- Prepare Observational Data: Assemble time-series or depth-profile data for state variables (e.g., CH4, acetate). Partition into calibration (e.g., 80%) and validation (20%) sets.
- Configure & Run MCMC: Initialize the sampler (e.g., No-U-Turn Sampler, NUTS) with multiple chains (e.g., 4). Run a sufficient number of iterations (e.g., 50,000) until convergence is diagnosed via R-hat (~1.01) and visual inspection of trace plots.
- Evaluate Posterior: Analyze the posterior distribution
P(θ | y_obs). Report the median and 95% credible intervals for each parameter. Compare prior vs. posterior to show data constraint. - Predictive Check: Use the posterior samples to run the model forward, generating a posterior predictive distribution. Plot this uncertainty band against the held-out validation data to assess predictive skill.
Diagram Title: Bayesian Calibration Process for Microbial Model Parameters
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Tools for Parameter Uncertainty Analysis in G2E Research
| Tool / Reagent | Category | Function in Analysis | Example/Note |
|---|---|---|---|
| SALib (Python) | Software Library | Implements Global Sensitivity Analysis methods (Sobol', Morris, FAST). | Enables efficient design and analysis of GSA. |
| PyMC / Stan | Software Library | Probabilistic programming frameworks for Bayesian calibration. | Uses MCMC or variational inference to sample posteriors. |
| High-Performance Computing (HPC) Cluster | Infrastructure | Manages thousands of model runs required for GSA and MCMC. | Cloud-based (AWS, GCP) or institutional clusters are essential. |
| Model Emulator (Surrogate) | Analytical Tool | A fast statistical approximation (e.g., Gaussian Process) of a slow process-based model. | Dramatically reduces computational cost of GSA and Bayesian inference. |
| Multi-Omics Datasets | Calibration Data | Provides priors and calibration targets (e.g., enzyme abundances, metatranscriptome). | Used to constrain Vmax, Km via relationships in likelihood function. |
| Ecosystem Flux Measurements | Validation Data | Independent data for posterior predictive checks (e.g., eddy covariance, soil chamber fluxes). | Validates the integrated model's real-world predictive skill. |
Within the Genome-to-Ecosystem (G2E) research framework, a critical bottleneck is mechanistically linking genomic potential to measurable ecosystem processes. Microbial traits—physiological, morphological, or life-history attributes—are the conceptual bridge. This application note details protocols for applying machine learning (ML) to discover and quantify hidden relationships between microbial traits (e.g., growth rate, enzyme affinity, stress resistance) and biogeochemical functions (e.g., CO2 flux, nitrification rate, lignin decay). By moving beyond correlation to predictive modeling, ML enables the parameterization of traits in ecosystem models, fulfilling a core objective of G2E integration.
Core Data & ML Approaches
The following table summarizes current ML applications in microbial trait-function discovery, based on recent literature.
Table 1: ML Models for Trait-Function Prediction in Microbial Systems
| ML Model Category | Example Algorithms | Typical Input Data | Predicted Trait/Function | Reported Performance (R²/Accuracy) | Key Advantage for G2E |
|---|---|---|---|---|---|
| Supervised Regression | Random Forest, Gradient Boosting, Neural Networks | Genomic features (e.g., KEGG/EC numbers, Pfam counts), Metatranscriptomics, Environmental metadata | Enzyme kinetics (Vmax, Km), Growth yield, Methane production rate, Organic matter decomposition rate | 0.65 - 0.89 (R²) for process rates | Handles high-dimensional, non-linear relationships; provides feature importance. |
| Dimensionality Reduction | t-SNE, UMAP, Autoencoders | Metagenome-assembled genomes (MAGs), Community metabolomics, Phenotypic arrays | Trait-based microbial guilds, Functional niche spaces | N/A (Visualization/Clustering) | Identifies latent ecological strategies and reduces redundancy for model input. |
| Integrative Networks | Graphical Models, Co-inertia Analysis | Multi-omics layers (Genome, Transcriptome, Proteome) coupled with process measurements | Causal links between gene abundance and process, e.g., nifH → N2 fixation | Edge accuracy > 0.80 in synthetic benchmarks | Infers putative mechanistic pathways for hypothesis generation. |
Detailed Application Notes & Protocols
Protocol: Predictive Modeling of Decomposition Rates from Genomic Traits
Objective: Train a model to predict litter decomposition rate (k) from the genomic trait profiles of a microbial community.
Materials & Workflow:
- Data Acquisition:
- Genomic Traits: From shotgun metagenomics of litter samples, calculate gene family abundances (e.g., CAZy for glycoside hydrolases, Peroxibase for peroxidases). Normalize as counts per million (CPM).
- Function Measurement: Experimentally determine decomposition rate k using litter bag techniques or respirometry (CO2 evolution). Express as % mass loss per day.
- Environmental Covariates: Record pH, moisture, C:N ratio, temperature.
Feature Engineering & Preprocessing:
- Perform log10(x+1) transformation on gene abundance data.
- Use variance thresholding to remove low-variance gene features (<1% variance).
- Standardize all features (genomic and environmental) using StandardScaler.
Model Training & Validation (Random Forest Regression):
- Split data (n samples) into training (70%) and hold-out test (30%) sets.
- Train a Random Forest Regressor (scikit-learn) on the training set. Use nested cross-validation (5-fold inner, 3-fold outer) for hyperparameter tuning (nestimators, maxdepth).
- Evaluate on the hold-out test set. Report R², Mean Absolute Error (MAE), and Root Mean Square Error (RMSE).
- Extract and plot feature importance (Gini importance or SHAP values) to identify top predictive genomic traits.
G2E Integration:
- The trained model serves as a "trait-to-function module." For novel metagenomes, input processed trait data to predict k.
- Sensitivity Analysis: Perturb key trait inputs in the model to simulate microbial community changes and forecast impacts on ecosystem carbon cycling.
ML Workflow for Predicting Decomposition Function
Protocol: Unsupervised Discovery of Trait-Based Guilds using Dimensionality Reduction
Objective: Identify coherent microbial functional groups (guilds) based on multi-trait profiles, independent of taxonomy.
Methodology:
- Trait Matrix Construction: For a set of MAGs, build a matrix where rows are genomes and columns are traits (e.g., presence of metabolic pathways, optimal growth pH, codon usage bias, rRNA copy number). Use binary (0/1) or continuous values.
- Dimensionality Reduction with UMAP:
- Apply UMAP (Uniform Manifold Approximation and Projection) using the
umap-learnPython library. - Parameters:
n_neighbors=15,min_dist=0.1,n_components=2, metric='jaccard'(for binary traits). - Fit and transform the trait matrix to obtain 2D coordinates for each MAG.
- Apply UMAP (Uniform Manifold Approximation and Projection) using the
- Cluster Identification: Apply HDBSCAN clustering on the UMAP embeddings. Use
min_cluster_size=5. MAGs not assigned to a cluster are labeled "noise." - Functional Guild Annotation: For each cluster, calculate the enrichment of specific traits (Fisher's exact test) and biogeochemical processes (e.g., denitrification steps). Define guilds by their shared trait suite (e.g., "high-affinity oligotrophs," "versatile fermentation specialists").
Discovery of Microbial Guilds via Trait Dimensionality Reduction
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for ML-Driven Trait-Function Research
| Item / Solution | Supplier Examples | Function in Protocol |
|---|---|---|
| ZymoBIOMICS DNA/RNA Miniprep Kits | Zymo Research | High-yield, inhibitor-free nucleic acid extraction from complex environmental samples for omics sequencing. |
| NEBNext Ultra II DNA Library Prep Kit | New England Biolabs | Preparation of sequencing-ready libraries from metagenomic DNA for trait gene profiling. |
| MiSeq/HiSeq & NovaSeq Systems | Illumina | Platform for high-throughput shotgun metagenomic and metatranscriptomic sequencing. |
| MicroResp or Phenotype MicroArrays | Biolog Inc. | High-throughput measurement of community-level physiological traits (substrate use). |
| QIIME 2, PICRUSt2, METABOLIC | Open Source BioBakery | Bioinformatics pipelines for processing sequence data into functional trait tables (KEGG, MetaCyc). |
| scikit-learn, XGBoost, PyTorch | Open Source (Python) | Core ML libraries for building, training, and evaluating predictive models. |
| SHAP (SHapley Additive exPlanations) | Open Source (Python) | Model interpretation tool to quantify the contribution of each genomic trait to a function prediction. |
| Google Colab Pro / AWS SageMaker | Google Cloud, Amazon Web Services | Cloud computing platforms with GPU access for running computationally intensive ML training. |
Within the broader thesis on the Genome-to-Ecosystem (G2E) framework, a central challenge is scaling microbial trait-based simulations to ecosystem-relevant scales. Traditional models fail to capture the high-resolution, spatially-explicit interactions between genetically encoded microbial traits (e.g., nutrient uptake kinetics, stress response) and heterogeneous environmental matrices (e.g., soil aggregates, root surfaces, aquatic microzones). This document details application notes and protocols for employing HPC solutions to overcome computational bottlenecks, enabling predictive, mechanistic G2E modeling that integrates omics-derived traits into biogeochemical fate and transport simulations.
Core Computational Challenges in Spatially-Explicit G2E
Table 1: Key Computational Bottlenecks and HPC Mitigation Strategies
| Bottleneck Category | Specific Challenge in G2E Simulations | HPC Solution Approach | Typical Performance Gain |
|---|---|---|---|
| Spatial Resolution | Simulating microbial communities at µm-mm scale across meter-km domains. | MPI-based domain decomposition; Adaptive Mesh Refinement (AMR). | 10-100x scaling on 100s-1000s of cores. |
| Agent/Individual-Based Complexity | Tracking traits, states, and interactions of 10^6-10^9 individual microbial agents. | Hybrid MPI+OpenMP/MPI+CUDA for agent kernels; efficient spatial indexing (e.g., k-d trees). | 50-200x faster agent processing. |
| Reaction-Transport Coupling | Solving coupled PDEs for biogeochemistry with stochastic trait-based microbial metabolism. | Operator splitting solved on separate compute partitions; GPU acceleration for reaction kernels. | 5-20x faster time-to-solution. |
| Parameter Uncertainty & Ensembles | Running 10^3-10^5 simulations for global sensitivity analysis (GSA) & calibration. | High-throughput job arrays on cluster schedulers (Slurm, PBS); workflow managers (Nextflow, Snakemake). | Linear scaling with allocated nodes. |
| Data I/O & In Situ Analysis | Writing/reading terabytes of spatiotemporal state data (e.g., 4D concentration fields). | Parallel I/O (e.g., HDF5, NetCDF-4); in situ visualization/analysis libraries (e.g., ParaView Catalyst). | I/O time reduced by 70-90%. |
Application Notes: Reference HPC Architecture and Software Stack
Table 2: Recommended HPC Stack for G2E Simulations
| Layer | Component | Recommended Options | Role in G2E Workflow |
|---|---|---|---|
| Hardware | Compute Nodes | CPU clusters (AMD EPYC, Intel Xeon) + GPU accelerators (NVIDIA A100, H100). | CPU for host logic, GPUs for parallelizable agent/RHS computations. |
| Parallelism | Programming Model | MPI (for inter-node), OpenMP/ CUDA (for intra-node). | Domain decomposition (MPI), thread-level parallelism on shared memory (OpenMP/CUDA). |
| Scheduler | Workload Manager | Slurm, PBS Pro, LSF. | Orchestrating ensemble runs, managing resource allocation. |
| Modeling Framework | Core Simulation Engine | Modified/configured versions of: Daisy (soil), IBMF (Individual-Based), PFLOTRAN (reactive transport), custom C++/Fortran+Python. | Solves the core spatially-explicit G2E model. |
| Pre/Post-Processing | Data & Workflow Tools | Snakemake/Nextflow (pipelines), Python (NumPy, SciPy, pandas), R. | Parameter generation, job submission, results aggregation. |
| Visualization | Analysis Suite | ParaView (parallel), VisIt, matplotlib (for 2D summaries). | Visualizing 3D/4D simulation outputs. |
Detailed Experimental Protocol: HPC-Enabled G2E Simulation Workflow
Protocol Title: Execution of a High-Resolution, Spatially-Explicit Microbial Nitrogen Cycling Simulation with Trait Variation.
Objective: To simulate the impact of genomic variation in amoA gene (encoding ammonia monooxygenase) kinetics on nitrification rates and N2O fluxes in a 3D soil core (1m x 1m x 0.5m) at 1mm resolution for 30 simulated days.
I. Pre-Simulation: Model Configuration & HPC Job Preparation
- Trait Parameterization:
- Input: Genomic data from metagenomes/isolates → amoA gene sequences.
- Action: Use tool
KBase(or local pipeline) to infer maximum enzymatic reaction rate (Vmax) and substrate affinity (Km) for ammonia oxidation for each unique gene variant. Populate a trait database file (traits.csv).
- Spatial Grid & Initialization:
- Input: 3D soil scan (X-ray CT) defining porosity and bulk density. Pre-processed biogeochemical initial conditions (NH4+, O2 profiles).
- Action: Convert scan to a structured grid. Use a Python script to stochastically inoculate microbial agents (ammonia-oxidizing archaea/bacteria) into grid voxels, assigning trait parameters from
traits.csvprobabilistically based on relative abundance data. - Output:
grid_geometry.bin,initial_conditions.h5,agent_locations.h5.
- HPC Job Script Generation:
- Write a Slurm submission script (
run_g2e.slurm) specifying:--nodes=32,--tasks-per-node=4,--cpus-per-task=8--time=24:00:00- Module loads (e.g.,
module load openmpi/4.1.5 hdf5/1.12.2) - Execution command:
mpirun -np 128 ./g2e_solver -input config.yaml
- Write a Slurm submission script (
II. Core Simulation Execution on HPC
- Job Submission & Monitoring:
sbatch run_g2e.slurm- Monitor via
squeue -j <jobid>. Check performance metrics (CPU/GPU utilization, memory) using cluster-specific tools (e.g.,ganglia,jobstats).
- In Situ Analysis (Optional):
- The simulation code is linked with the ParaView Catalyst library. Every 1000 simulation timesteps, a Catalyst script extracts a 2D slice and computes summary statistics (total biomass, mean reaction rate), writing a lightweight
.csvfile without halting the main simulation.
- The simulation code is linked with the ParaView Catalyst library. Every 1000 simulation timesteps, a Catalyst script extracts a 2D slice and computes summary statistics (total biomass, mean reaction rate), writing a lightweight
III. Post-Simulation: Data Reduction and Analysis
- Data Aggregation:
- After job completion, the output is a series of parallel HDF5 files (
output_*.h5) per snapshot. - Use a post-processing script with parallel HDF5 to compute spatial integrals and time-series of key variables (e.g., total N2O flux, spatial variance of NH4+).
- After job completion, the output is a series of parallel HDF5 files (
- Ensemble Analysis (If Applicable):
- For parameter ensembles, a Python script using
Snakemakecollates results from multiple job directories into a single Pandas DataFrame for statistical analysis and visualization.
- For parameter ensembles, a Python script using
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Computational & Data "Reagents" for HPC G2E Research
| Item / Solution | Function in HPC G2E Research | Example Source / Specification |
|---|---|---|
| MPI Library | Enables distributed memory parallelism across compute nodes. | OpenMPI, MPICH, Intel MPI. |
| Parallel I/O Library | Manages efficient reading/writing of large simulation state files from multiple processes. | HDF5 with parallel enabled, NetCDF-4. |
| Performance Profiler | Identifies computational hotspots and load imbalances in the simulation code. | Intel VTune, NVIDIA Nsight Systems, HPCToolkit. |
| Workflow Manager | Automates and reproduces complex pipelines of preprocessing, simulation, and analysis. | Snakemake, Nextflow, Apache Airflow. |
| Container Platform | Ensures software environment portability and reproducibility across HPC systems. | Apptainer/Singularity, Docker (where supported). |
| Version Control System | Tracks changes to simulation code, configuration files, and analysis scripts. | Git, hosted on GitHub or GitLab. |
| Numerical Library | Provides optimized, parallelized routines for linear algebra and solvers. | PETSc, Intel MKL, CUDA-enabled libraries. |
Visualizations
Diagram 1: HPC G2E Simulation Software Stack Architecture
Diagram 2: Protocol for Spatially-Explicit G2E Simulation on HPC
Proving the Paradigm: Validation Strategies and Comparative Advantages of G2E Models
Within the Genome-to-Ecosystem (G2E) research framework, a central challenge is empirically validating model predictions that link genomic potential to biogeochemical function. This requires moving beyond correlation to establish causal, mechanistic links between microbial traits and ecosystem-scale processes. Stable Isotope Probing (SIP) combined with controlled microcosm experiments provides a critical validation benchmark. These methods allow researchers to trace the incorporation of substrates into specific microbial taxa and their biomolecules, directly testing hypotheses about functional guilds, metabolic pathways, and turnover rates predicted from genomic data. This protocol details the integrated application of SIP-microcosm experiments for validating G2E model outputs.
Key Application Notes
Role in the G2E Workflow
SIP-microcosm experiments act as a crucial intermediary validation step. Genomic and metagenomic data predict potential functions (e.g., presence of amoA genes for nitrification). Process models make predictions about rates under environmental conditions. SIP-microcosm experiments test these predictions by directly identifying the active taxa performing the function and measuring the process rate under defined conditions, thereby closing the loop between gene and ecosystem.
Selection of Isotopic Tracer
The choice of isotope (¹³C, ¹⁵N, ¹⁸O, ²H) and its molecular form is dictated by the target biogeochemical process and the genomic prediction being tested. For instance, ¹³C-labeled methane tests predictions about methanotroph identity and activity in a soil carbon model.
Critical Considerations
- Labeling Time Course: Must be optimized to capture active consumers without secondary labeling via trophic interactions.
- Isotope Enrichment Level: Typically 5-30 atom% excess, balancing cost and detection sensitivity (NanoSIMS requires lower enrichment than GC/MS).
- Microcosm Design: Must replicate key environmental conditions (e.g., redox, moisture, temperature) to ensure ecological relevance.
- Extraction Efficiency: Critical for nucleic acid-SIP (NA-SIP) or phospholipid fatty acid-SIP (PLFA-SIP) to avoid bias.
Experimental Protocols
Protocol: Coupled¹⁵N-Ammonia Oxidation Microcosm and DNA-SIP for Nitrifier Validation
Objective: To validate genomic predictions of ammonia-oxidizing archaea (AOA) vs. bacteria (AOB) activity in agricultural soil.
Materials:
- Soil cores from target site.
¹⁵NH₄Cl(99 atom%¹⁵N).- Serum bottles (120 mL) or custom microcosm chambers.
- GC-MS or IRMS for
¹⁵N-N₂O/NO₃analysis. - Ultracentrifuge and ultracentrifuge tubes for CsCl gradients.
- Lysis buffers, PCR reagents, and sequencing primers for amoA genes.
Procedure:
- Microcosm Setup: Homogenize soil under sterile conditions. Distribute 10g (wet weight) into 12 replicate serum bottles.
- Tracer Addition: To 6 bottles, add
¹⁵NH₄Clsolution (final concentration 50 µg N/g soil). To 6 control bottles, add equivalent¹⁴NH₄Cl. - Incubation: Incubate in the dark at in situ temperature. Sacrifice triplicate
¹⁵Nand¹⁴Nmicrocosms at time points T=0, T=24h, T=168h. - Process Rate Measurement: At each time point, extract soil with 2M KCl. Analyze
¹⁵Nenrichment inNO₃/NO₂pool via derivatization and GC-MS to calculate nitrification rate. - Nucleic Acid Extraction: Extract total DNA from all samples using a commercial soil DNA kit.
- Isopycnic Centrifugation: Prepare CsCl density gradient (1.72 g/mL average density) with 1 µg DNA. Ultracentrifuge at 176,000 x g, 20°C for 36-48h.
- Fractionation: Fractionate gradient into 12-14 fractions. Measure density via refractometry.
- Quantitative Analysis: Perform qPCR for AOA and AOB amoA genes on all fractions. Identify "heavy" (
¹⁵N-enriched) DNA fractions. - Sequencing & Analysis: Sequence amoA amplicons from "heavy" (
¹⁵N) and "light" (¹⁴Ncontrol) fractions. Compare AOA/AOB community structure. Taxa enriched in the "heavy" fraction of¹⁵Ntreatments are active ammonia oxidizers.
Protocol:¹³C-Cellulose Degradation and RNA-SIP for Active Degrader Identification
Objective: Identify active cellulose-degrading fungi and bacteria predicted from metagenome-assembled genomes (MAGs).
Materials:
¹³C-labeled cellulose (e.g.,U-¹³Ccellulose).¹²C-cellulose control.- RNA extraction kit (with bead-beating).
- Cesium trifluoroacetate (CsTFA) for RNA gradients.
- Reverse transcription and RT-qPCR reagents.
- cDNA sequencing library prep kit.
Procedure:
- Substrate Addition: Add
¹³C- or¹²C-cellulose (1% w/w) to triplicate soil microcosms. - Incubation: Incubate, measuring
¹³CO₂evolution via cavity ring-down spectroscopy. - RNA Extraction: Extract total RNA at peak
CO₂evolution. Treat with DNase. - RNA-SIP Density Gradient: Use CsTFA for isopycnic centrifugation of RNA (e.g., 169,000 x g, 36h, 20°C).
- Fractionation & Analysis: Fractionate, precipitate RNA, and convert to cDNA.
- Functional Gene Profiling: Perform RT-qPCR for glycoside hydrolase family 48 (cellulase) genes on all fractions.
- Metatranscriptomics: Sequence cDNA from heavy (
¹³C) and light (¹²C) fractions. Map transcripts to MAGs from the same system. Active degraders show transcript enrichment in the heavy fraction.
Data Presentation
Table 1: Example SIP-Microcosm Data Output for Nitrification Validation
| Microcosm Treatment | Incubation Time (h) | Nitrification Rate (µg N g⁻¹ day⁻¹) | AOA amoA ¹⁵N-Heavy Fraction Copy Number (x10⁸ g⁻¹) |
AOB amoA ¹⁵N-Heavy Fraction Copy Number (x10⁸ g⁻¹) |
Dominant Active Taxa (Heavy Fraction) |
|---|---|---|---|---|---|
¹⁵NH₄⁺ |
0 | 0.0 ± 0.0 | 0.01 ± 0.00 | 0.01 ± 0.00 | N/A |
¹⁵NH₄⁺ |
24 | 1.8 ± 0.2 | 5.2 ± 0.8 | 0.3 ± 0.1 | Nitrososphaera spp. (AOA) |
¹⁵NH₄⁺ |
168 | 0.5 ± 0.1 | 8.1 ± 1.2 | 2.4 ± 0.5 | Nitrososphaera & Nitrosospira |
¹⁴NH₄⁺ (Control) |
168 | 1.9 ± 0.3 | 0.02 ± 0.01 | 0.01 ± 0.00 | N/A |
Table 2: Key Research Reagent Solutions for SIP-Microcosm Validation
| Item | Function in Validation Experiment | Example Product/Specification |
|---|---|---|
¹³C/¹⁵N-Labeled Substrates |
Tracer for linking specific metabolic activity to organism identity. | ¹³C-CH₄ (99%), ¹⁵N-NH₄Cl (99%), ¹³C-Cellulose (U-¹³C, 98%). |
| CsCl / CsTFA, UltraPure | Forms density gradient for separation of "heavy" labeled biomolecules. | Density gradient grade, for molecular biology. |
| Ultracentrifuge & Tubes | Essential for isopycnic centrifugation in SIP. | Fixed-angle or near-vertical rotors; thick-walled polyallomer tubes. |
| Soil DNA/RNA Shield & Kits | Preserves in situ transcriptome and enables efficient nucleic acid extraction from complex matrices. | Bead-beating based kits optimized for humic acid removal. |
| Density Fractionation System | Precisely collects density gradient fractions for downstream analysis. | Piston gradient fractionator or automated pipetting system. |
| Isotope-Ratio MS (IRMS) or GC-MS | Precisely measures isotopic enrichment in gases, solutes, or biomarkers (PLFAs). | Coupled to automated sample preparation interfaces (e.g., gas bench, precon). |
| Taxon/Function-Specific qPCR Assays | Quantifies target genes in density fractions to identify "heavy" nucleic acids. | Validated primer-probe sets for amoA, mcrA, rbcL, etc. |
| NanoSIMS-Compatible Carriers | Allows spatially-resolved SIP at the single-cell level (advanced application). | Conductive, epoxy-based embedding resins. |
Mandatory Visualizations
Title: SIP-Microcosm Validation Workflow in G2E Research
Title: Principle of Stable Isotope Probing (SIP)
This application note is framed within a broader thesis on the Genome-to-Ecosystem (G2E) framework, which aims to integrate microbial genomic traits and community dynamics into predictive biogeochemical models. The objective is to compare the predictive accuracy and mechanistic insight of emerging G2E models against established traditional stoichiometric models for nitrogen cycling processes (e.g., nitrification, denitrification, N-fixation).
Table 1: Model Performance Comparison for Predicting Net Nitrification Rates
| Model Class | Specific Model Name/Type | R² (Range) | RMSE (mg N kg⁻¹ day⁻¹) | Key Predictor Variables | Spatial Scale Tested |
|---|---|---|---|---|---|
| Traditional Stoichiometric | CENTURY/DAYCENT | 0.45 - 0.65 | 0.15 - 0.35 | Soil C:N, pH, Temperature, Moisture, Bulk N Pool | Plot to Regional |
| Traditional Stoichiometric | DNDC | 0.50 - 0.70 | 0.12 - 0.30 | Soil Texture, Climate, Fertilizer Input, Crop Type | Field to Regional |
| G2E Framework | MEND (Microbial-ENzyme) | 0.65 - 0.85 | 0.08 - 0.20 | amoA Gene Abundance, Enzyme Vmax/Km, Microbial C:N, EPS | Microcosm to Watershed |
| G2E Framework | DEMENT (DEcomposition Microbial-Explicit Theory) | 0.70 - 0.88 | 0.07 - 0.18 | Microbial rRNA Operon Copy Number, Genomic POT/NasA Traits, Community Structure | Lab Incubation to Ecosystem |
Table 2: Key Differences in Model Structure and Data Requirements
| Feature | Traditional Stoichiometric Models | G2E Models |
|---|---|---|
| Core Unit | Bulk Nutrient Pools (e.g., NH₄⁺, NO₃⁻) | Microbial Functional Groups / Genomic Traits |
| Rate Formulation | Empirical or Michaelis-Menten, abiotic drivers dominant | Mechanistic, microbially-mediated, trait-based parameters |
| Nitrogen Process Links | Often decoupled or linear | Tightly coupled via microbial biomass & energy constraints |
| Key Data Inputs | Soil chemistry, climate, vegetation type | Metagenomes, metatranscriptomes, enzyme assays, PLFAs |
| Temporal Resolution | Daily to Yearly | Hourly to Daily |
| Computational Demand | Low to Moderate | High (requires genomic & community data assimilation) |
Experimental Protocols
Protocol 1: Establishing a Model Benchmarking Experiment
Title: In-Situ Measurement of Nitrification Rates for Model Validation Purpose: To generate empirical data on gross and net nitrification rates across gradients for validating G2E and traditional models. Materials: See "Scientist's Toolkit" below. Procedure:
- Site Selection: Identify 10-20 study plots representing a gradient of soil C:N (e.g., 10-30), pH (5-8), and land use.
- Core Collection: Collect triplicate soil cores (0-15 cm depth) from each plot using sterile corers. Process immediately or store at 4°C for <24h.
- ¹⁵N Isotope Pool Dilution (Gross Rates): a. For each core, prepare two sets of subsamples (20g fresh weight). b. Inject one set with (¹⁵NH₄)₂SO₄ solution and the other with K¹⁵NO₃ solution to achieve 5-10 at% enrichment. c. Incubate in the dark at in-situ temperature. Sacrifice replicates at T=0, 6, 24, and 48 hours. d. Extract NH₄⁺ and NO₃⁻ with 2M KCl. Filter extracts. e. Analyze isotopic composition of NH₄⁺ and NO₃⁻ via diffusion coupled to Isotope Ratio Mass Spectrometry (IRMS). f. Calculate gross nitrification (production of NO₃⁻ from NH₄⁺) and mineralization rates using isotope mixing models.
- Net Rate Incubation: a. Incubate additional intact cores (sieved, 2mm) aerobically for 14 days at field moisture capacity. b. Extract NH₄⁺ and NO₃⁻ at days 0, 7, and 14 via 2M KCl. c. Analyze concentrations via colorimetric continuous flow analyzer. d. Calculate net nitrification rate as linear accumulation of NO₃⁻ over time.
- Ancillary Data Collection: Measure soil pH, total C/N (Elemental Analyzer), moisture, texture. Preserve soil aliquots at -80°C for genomic analysis.
Protocol 2: Parameterizing a G2E Model (MEND Framework)
Title: Acquisition of Microbial Trait Parameters for G2E Model Input Purpose: To generate direct inputs for a G2E model from soil samples. Procedure:
- Nucleic Acid Extraction: Extract total DNA and RNA from 0.5g of frozen soil using a commercial kit optimized for environmental samples (e.g., DNeasy PowerSoil Pro, RNeasy PowerSoil Total RNA Kit). Include DNase treatment for RNA extracts.
- Quantitative PCR (qPCR) for Functional Genes: a. Design/use primers for key N-cycling genes: bacterial & archaeal amoA (nitrification), nirK, nirS, nosZ (denitrification). b. Prepare standard curves from cloned gene fragments of known concentration. c. Perform triplicate qPCR reactions on DNA extracts using a SYBR Green master mix. d. Calculate gene abundances per gram dry soil.
- Metagenomic Sequencing & Trait Inference: a. Prepare sequencing libraries from DNA extracts (e.g., Illumina NovaSeq, 150bp paired-end). b. Process reads: quality filter, assemble (co-assembly per site recommended), predict genes. c. Annotate genes against functional databases (KEGG, UniRef). d. Extract trait proxies: average 16S rRNA gene copy number (from rrnDB), presence/abundance of high-affinity vs. low-affinity enzyme variants (e.g., amoCAB clusters), genomic nitrogen content estimates from protein sequences.
- Enzyme Activity Kinetics: a. Measure potential activities of enzymes: Ammonia Monooxygenase (AMO) via substrate-induced respiration inhibition, Nitrite Oxidoreductase (NXR). b. Perform substrate saturation curves to estimate Vmax and Km for key enzymes (e.g., using chlorate inhibition for AMO).
- Model Integration: Compile trait data (gene abundances, Vmax/Km, community-weighted genomic traits) into the microbial explicit functional parameters of the G2E model structure.
Diagrams
Diagram 1: Conceptual Workflow for the Comparative Analysis
Title: Workflow for G2E vs. Traditional Model Comparison
Diagram 2: Structural Comparison of Model Approaches
Title: Structural Differences Between Model Classes
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Nitrogen Cycling Model Benchmarking
| Item Name / Reagent | Function / Application | Example Product / Specification |
|---|---|---|
| Isotope Tracers | Labeling NH₄⁺ and NO₃⁻ pools for measuring gross N transformation rates via isotope dilution. | (¹⁵NH₄)₂SO₄ (98 at% ¹⁵N), K¹⁵NO₃ (98 at% ¹⁵N); Cambridge Isotope Laboratories. |
| Soil DNA/RNA Kit | Simultaneous or separate isolation of high-quality, inhibitor-free nucleic acids from complex soil matrices. | DNeasy PowerSoil Pro Kit (Qiagen), RNeasy PowerSoil Total RNA Kit (Qiagen). |
| qPCR Master Mix | Sensitive detection and quantification of functional gene abundances from environmental DNA extracts. | SYBR Green PCR Master Mix (Thermo Fisher), with optimized buffers for inhibitor-prone samples. |
| N Analysis Consumables | For colorimetric determination of NH₄⁺ and NO₃⁻ concentrations in soil extracts. | Seal Analytical AA3 HR Continuous Flow Analyzer reagents or equivalent microplate assay kits. |
| Enzyme Substrates | To measure potential enzyme activities (e.g., AMO, NIR, NOS) for kinetic parameter estimation. | Sodium chlorate (AMO inhibitor), acetylene (NOS inhibitor), specific fluorogenic substrates. |
| Bioinformatics Pipeline | For processing metagenomic data to extract microbial trait information. | Software: Trimmomatic, MEGAHIT, Prokka, HUMAnN. Run on HPC or cloud (Google Cloud, AWS). |
| Modeling Software | Platforms for building and running the biogeochemical models. | R/Python with packages (deSolve, FME) for custom models; pre-built model code (MEND, DNDC 95). |
Within the Genome-to-Ecosystem (G2E) framework, predicting microbial community responses to novel perturbations is the ultimate test of model integration. This analysis evaluates the predictive power of different modeling approaches—from trait-based to genome-informed dynamic models—when forecasting community dynamics and biogeochemical outcomes under antibiotic stress, a common and clinically relevant perturbation.
Quantitative Comparison of Model Predictive Performance
Table 1: Predictive Accuracy of Models for Antibiotic Perturbation Outcomes
| Model Class | Key Inputs | Prediction Target | Reported R² / Accuracy | Major Limitation | Reference (Year) |
|---|---|---|---|---|---|
| Statistical (ML) | 16S rRNA amplicon data, antibiotic metadata | Species abundance shifts | 0.65-0.78 (R²) | Poor extrapolation beyond training data | Recent (2023) |
| Consumer-Resource Model (CRM) | Genomically-inferred metabolic traits, resource supply | Community composition & metabolite fluxes | 0.70-0.82 (R² for abundance) | Requires precise resource uptake parameters | Recent (2024) |
| Dynamic Energy Budget (DEB) | Genomic size, rRNA operon count, antibiotic MIC | Biomass yield & respiration under stress | 0.75-0.85 (R² for growth rate) | Computationally intensive | Recent (2023) |
| Genome-Scale Metabolic Modeling (GEM) | Annotated genomes, transport reactions | Cross-feeding resilience & community productivity | 0.60-0.75 (F1-score for survival) | Misses ecological interactions | Recent (2024) |
| Integrated G2E Hybrid | GEMs + trait-mediated interaction parameters | Ecosystem function (e.g., nitrification rate) | 0.80-0.90 (R² for function) | High data requirement, complex calibration | Current Thesis |
Table 2: Key Traits for Predicting Antibiotic Response in a G2E Context
| Trait Category | Specific Trait | Measurement/Proxy | Influence on Ecosystem Function Post-Perturbation |
|---|---|---|---|
| Resistance | Antibiotic Minimum Inhibitory Concentration (MIC) | Broth microdilution assay; genomic resistance gene presence | Direct survival; determines initial biomass loss |
| Tolerance | Lag time extension, death rate | Growth curve analysis under stress | Modifies biogeochemical process rates during stress period |
| Metabolic Flexibility | Number of alternative carbon utilization pathways | pangenome analysis; flux balance analysis plasticity | Recovery rate of community-level respiration post-antibiotic |
| Interaction Strength | Cross-feeding dependency (obligate/facultative) | Metabolite exchange network from GEMs | Resilience of community structure; prevents collapse |
| Stress-Induced Secretion | Public good (e.g., siderophore) production rate | Reporter assays; genomic biosynthetic cluster identification | Maintains community function via cooperative behavior |
Experimental Protocols for Ground-Truthing Predictions
Protocol 3.1: Controlled Perturbation Microcosm for G2E Validation
Objective: Generate empirical data on community structural and functional response to antibiotics to validate G2E model predictions. Materials: Defined microbial community, modified M9 or soil extract medium, antibiotic stock, bioreactors (e.g., BioLector), LC-MS/MS, Illumina MiSeq.
- Inoculum Preparation: Grow defined bacterial isolates to mid-log phase. Mix to form a defined consortium with known genomic and trait data.
- Perturbation Setup: In a 48-well microtiter plate bioreactor, add 1.5 mL medium per well. Inoculate at a standardized OD600. Apply a gradient of antibiotic concentration (0, 0.5x, 1x, 2x MIC of keystone species).
- High-Resolution Monitoring: Incubate with continuous monitoring of OD600 (biomass), pH, and dissolved O₂. Sample every 2 hours for 48h.
- Endpoint Analyses: At 48h, extract DNA for 16S rRNA gene amplicon sequencing (V4 region). Filter supernatant for extracellular metabolite analysis via LC-MS/MS. Measure a key ecosystem process (e.g., nitrate concentration via colorimetric assay).
- Data Integration: Correlate shifts in relative abundance with trait database (e.g., MIC, genome size). Fit process rates to DEB or CRM models.
Protocol 3.2: Trait-Based Model Calibration Using Phenotypic Microarrays
Objective: Measure microbial growth phenotypes under stress to parameterize trait-based models. Materials: BIOLOG GEN III plates or custom phenotype microarray, isolated strains, antibiotic, plate reader.
- Plate Preparation: Supplement BIOLOG PM plates with a sub-inhibitory concentration of antibiotic (e.g., 0.25x MIC) in test wells. Use untreated control plates.
- Inoculation: Suspend washed microbial cells in inoculating fluid. Dispense 100 µL per well.
- Incubation and Reading: Incubate at appropriate temperature. Measure absorbance at 590 nm every 15 minutes for 72h using a kinetic plate reader.
- Data Analysis: Calculate area under the curve (AUC) for each carbon source. Derive traits: specific growth rate, lag time, and substrate utilization versatility index under stress. Input as species parameters into a consumer-resource model.
Visualization of Conceptual Frameworks and Workflows
Diagram 1: G2E Predictive Framework for Novel Perturbations (100 chars)
Diagram 2: Validation Workflow for Antibiotic Perturbation (90 chars)
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Materials for G2E Perturbation Studies
| Item Name | Supplier Examples | Function in Experiment |
|---|---|---|
| Defined Microbial Community | ATCC, DSMZ | Provides known genomic background for trait-based prediction; reduces complexity. |
| Biolog Phenotype Microarray Plates | Biolog Inc. | High-throughput profiling of metabolic traits under stress for model parameterization. |
| BioLector Microbioreactor System | m2p-labs | Enables parallel, online monitoring of biomass and pH in 48-96 parallel microcosms. |
| ZymoBIOMICS Spike-in Control | Zymo Research | Internal standard for metagenomic sequencing to quantify absolute abundance shifts. |
| Tetracycline Hydrochloride (or other antibiotics) | Sigma-Aldrich | Standardized perturbation agent; used in gradient to test model extrapolation. |
| DNeasy PowerSoil Pro Kit | Qiagen | Robust DNA extraction from diverse, possibly lysed, communities post-antibiotic. |
| KEGG & ModelSEED Databases | Public Access | For genome annotation and constructing genome-scale metabolic models (GEMs). |
| Microbial Trait Database (MiTRA) | Public Database | Curated repository of microbial traits (e.g., growth rate, optimal pH) for priors. |
| COMETS Python Platform | Public Software | Simulates dynamic metabolism of microbial communities using GEMs in space & time. |
In the context of a Genome-to-Ecosystem (G2E) framework, which integrates microbial trait data derived from genomic information into predictive biogeochemical models, quantifying model improvement is paramount. This integration aims to enhance the prediction of ecosystem-scale processes, such as carbon sequestration, nitrogen cycling, and methane emission. For researchers, scientists, and drug development professionals exploring microbial interventions for climate mitigation or bioprospecting, rigorous evaluation of these multi-scale models is essential. This document outlines standardized metrics and experimental protocols for assessing model performance across the critical axes of accuracy, robustness, and generality, ensuring that improvements in G2E models translate to reliable, actionable insights.
Part 1: Core Evaluation Metrics
The performance of a G2E model must be assessed using a suite of complementary metrics. The following tables summarize key quantitative measures for each evaluation pillar.
Table 1: Metrics for Model Accuracy (Predictive Performance)
| Metric | Formula/Description | Application in G2E Context |
|---|---|---|
| Root Mean Square Error (RMSE) | √[Σ(Pᵢ - Oᵢ)²/n] | Quantifies average error in predicting a continuous biogeochemical flux (e.g., CO₂ emission rate). Lower values indicate better fit. |
| Normalized RMSE (NRMSE) | RMSE / (Omax - Omin) | Allows comparison of error magnitude across different ecosystem variables (e.g., N₂O vs. CH₄ fluxes). |
| Coefficient of Determination (R²) | 1 - [Σ(Pᵢ - Oᵢ)² / Σ(Oᵢ - Ō)²] | Proportion of variance in observed ecosystem data explained by the model. Target: >0.6 for credible mechanistic insight. |
| Mean Absolute Error (MAE) | Σ|Pᵢ - Oᵢ| / n | Robust to outliers; useful for assessing typical deviation in predicted microbial growth rates or substrate uptake. |
| Probability of Detection (POD) | Hits / (Hits + Misses) | For binary events (e.g., methanogenesis threshold crossed). Evaluates model's ability to detect an observed event. |
| False Alarm Ratio (FAR) | False Alarms / (Hits + False Alarms) | Measures the fraction of predicted events that did not occur. Balances POD assessment. |
Table 2: Metrics for Model Robustness (Stability & Uncertainty)
| Metric | Formula/Description | Application in G2E Context |
|---|---|---|
| Sensitivity Index (Sᵢ) | (ΔY/Y) / (ΔXᵢ/Xᵢ) | Measures relative change in a key output (Y, e.g., net primary production) given a perturbation to parameter Xᵢ (e.g., microbial mortality rate). |
| Coefficient of Variation (CV) of Predictions | (σpred / μpred) * 100% | Assesses prediction stability across multiple bootstrap or cross-validation runs. Lower CV indicates higher robustness. |
| 95% Confidence Interval Width | Q97.5 - Q2.5 of posterior predictive distribution | Width of the uncertainty band around a prediction (e.g., soil respiration forecast). Narrower intervals denote higher confidence. |
| Parameter Identifiability | Ranks from posterior diagnostics (e.g., R-hat ~1.0) | In Bayesian calibration, indicates whether microbial trait parameters (e.g., substrate affinity) are well-constrained by data. |
Table 3: Metrics for Model Generality (Transferability)
| Metric | Formula/Description | Application in G2E Context |
|---|---|---|
| Spatial Transfer Error | RMSEtestsite / RMSEtrainsite | Performance loss when a model calibrated on one ecosystem (e.g., temperate forest) is applied to another (e.g., tropical grassland). |
| Temporal Transfer Error | RMSEfutureperiod / RMSEcalibrationperiod | Performance loss when projecting beyond the calibration period under climate change scenarios. |
| Process Generalization Index | Correlation(Ppred, Pobs) for a novel process | Ability to predict a related but untrained process (e.g., model trained on C cycling predicts N mineralization). |
| Trait-Informed vs. Statistic Benchmark | (Perftraitmodel - Perfstatmodel) / Perfstatmodel | Relative improvement of a mechanistic, trait-based G2E model over a purely statistical or phenomenological baseline. |
Part 2: Experimental Protocols
Protocol 1: Calibration and Accuracy Assessment of a Microbial-Enzyme G2E Model
Objective: To calibrate a model linking genomic potential for enzyme production to ecosystem-scale litter decomposition rates and quantify its accuracy. Materials: See "The Scientist's Toolkit" below. Procedure:
- Data Curation: Assemble paired datasets: (a) metagenomic/transcriptomic data quantifying gene abundances for key enzymes (e.g., glycoside hydrolases, peroxidases) from soil samples, and (b) concurrent in-situ measurements of litter mass loss and CO₂ efflux.
- Trait Parameterization: Derive microbial community-aggregated traits (e.g., maximum catalytic rate
k_cat, enzyme half-saturationK_m) from genomic data using predefined genomic-to-trait mapping databases. - Model Calibration: Implement a Markov Chain Monte Carlo (MCMC) algorithm to calibrate unknown scaling parameters (ξ) in the model
Decomp = f([Enzyme], Trait_k_cat, Trait_K_m; ξ)against observed decomposition rates. - Accuracy Quantification: Withhold 30% of site-year data as a validation set. Run the calibrated model and compute metrics from Table 1 (RMSE, R², MAE) comparing predictions to validation observations.
- Reporting: Document final parameter values, convergence diagnostics, and a table of validation metrics.
Protocol 2: Perturbation Analysis for Robustness Evaluation
Objective: To assess model robustness to variations in input data and parameter values. Procedure:
- Input Data Bootstrapping: Resample (with replacement) the underlying genomic and environmental driver dataset 100 times. For each bootstrap sample, recalibrate the model.
- Prediction Stability Analysis: For a fixed future climate scenario, run each of the 100 calibrated models. For each output variable, calculate the mean and Coefficient of Variation (CV) across the 100 runs (Table 2).
- Global Sensitivity Analysis (GSA): Using a Latin Hypercube Sampling design, vary all key microbial trait parameters simultaneously within biologically plausible ranges. Run the model for each parameter set.
- Sensitivity Metric Calculation: Perform a multiple linear regression between varied parameters and model outputs. Calculate normalized sensitivity indices (Sᵢ) as standardized regression coefficients. Rank parameters by |Sᵢ|.
Protocol 3: Cross-Biome Validation for Generality Testing
Objective: To evaluate model transferability across distinct ecosystem types. Procedure:
- Site Selection: Identify three distinct biome types (e.g., Boreal Forest, Arid Grassland, Tropical Wetland) with available paired genomic and biogeochemical data.
- Training and Testing: Calibrate the model exhaustively on data from one biome (Training Biome). Apply the calibrated model without any further adjustment to the other two biomes (Test Biomes 1 & 2).
- Generality Metrics Calculation: For each test biome, compute the Spatial Transfer Error (Table 3). Compare performance to a null benchmark model (e.g., a linear regression using only climate variables).
- Trait-Based Diagnosis: Analyze failures in transfer by examining if microbial trait distributions in the test biomes fall outside the ranges represented in the training data.
Part 3: Visualizations
Title: G2E Model Development and Evaluation Workflow
Title: Three Protocols Link to Core Metric Pillars
Part 4: The Scientist's Toolkit
Table 4: Essential Research Reagent Solutions for G2E Model Evaluation
| Item/Reagent | Function in G2E Evaluation |
|---|---|
| Curated Genomic-to-Trait Databases (e.g., METAGENOTE, FAPROTAX, Traitar) | Map gene abundances to inferred microbial phenotypic traits (e.g., metabolic pathways, enzyme kinetics) for model parameterization. |
| Biogeochemical Reference Datasets (e.g., NEON, FLUXNET, ISRaD) | Provide standardized, high-quality observational data for model calibration and validation across diverse ecosystems. |
| Bayesian Calibration Software (e.g., Stan, PyMC3, MCMCpack) | Enable robust parameter estimation and uncertainty quantification through probabilistic model-data fusion. |
| Global Sensitivity Analysis Libraries (e.g., SALib, R sensitivity package) | Facilitate systematic perturbation of model parameters to identify key drivers and assess robustness. |
| High-Performance Computing (HPC) Cluster Access | Provides necessary computational resources for running ensemble model simulations, bootstrapping, and complex MCMC calibrations. |
| Containerization Software (Docker/Singularity) | Ensures reproducibility of model evaluation workflows by encapsulating the exact software environment and dependencies. |
Community Standards and Shared Repositories for Model Validation and Reproducibility
Within the Genome-to-ecosystem (G2E) framework, microbial trait data must be integrated into predictive biogeochemical models to forecast ecosystem responses. This integration faces significant challenges in reproducibility and validation due to heterogeneous data sources, inconsistent model parameterization, and disparate computational environments. Establishing community standards and utilizing shared repositories are critical for creating transparent, comparable, and reproducible workflows from genomic prediction to ecosystem-scale simulation.
Current Landscape & Quantitative Data
Table 1: Prevalence of Reproducibility Practices in Microbial Ecology & Biogeochemical Modeling (2022-2024 Survey Data)
| Practice | Adoption Rate (%) | Primary Cited Barrier |
|---|---|---|
| Public deposition of raw sequencing data (e.g., SRA) | 94% | None (Journal mandate) |
| Public deposition of code/scripts | 58% | Lack of time for cleaning/documentation |
| Use of version control (e.g., Git) | 67% | Steep learning curve |
| Use of containerization (e.g., Docker, Singularity) | 41% | Technical complexity |
| Provision of explicit, executable model workflows | 35% | Intellectual property concerns |
| Publication of model code with parameters | 52% | Use of proprietary software/platforms |
| Use of community-standard ontology (e.g., ENVO, ChEBI) | 49% | Ontology complexity/fragmentation |
Table 2: Major Public Repositories for G2E-Relevant Data & Models
| Repository Name | Primary Content Type | Key Features for Reproducibility |
|---|---|---|
| NCBI Sequence Read Archive (SRA) | Raw genomic/transcriptomic reads | Stable identifiers, standardized metadata fields |
| JGI Genome Portal | Assembled genomes & annotations | Integrated analysis tools, project-based data |
| ESS-DIVE | Environmental system science data | Emphasis on biogeochemical & field data |
| Zenodo | General-purpose (code, data, models) | DOIs, versioning, links to GitHub |
| BioModels | Curated computational models (SBML) | Model annotation, simulation reproducibility |
| Code Ocean | Executable code capsules | Cloud-based compute environment |
Community Standards: Protocols and Application Notes
Protocol: Standardized Metadata Reporting for Microbial Trait Experiments
Purpose: To ensure experimental data linking microbial genotypes to phenotypes (e.g., growth rate, substrate affinity) can be unambiguously interpreted and reused in trait-based models.
Materials:
- Cultured microbial strain(s).
- Growth medium components.
- Bioreactor or microplate reader.
- Relevant analytical instruments (HPLC, spectrophometer).
- Data logging software.
Procedure:
- Pre-experiment Documentation:
- Assign a persistent identifier (e.g., DOI, strain catalog number) to the microbial strain.
- Document all growth medium components using community ontologies (e.g., ChEBI for chemical entities). Specify exact concentrations, pH, and ionic strength.
- Document environmental conditions: temperature, agitation, light (if relevant), and bioreactor vessel geometry.
- Describe the measurement technology (e.g., optical density at specified wavelength, substrate consumption rate) with instrument model and calibration method.
- Data Collection:
- Record time-series data in a non-proprietary format (e.g., .csv, .tsv).
- Include raw instrument readings alongside any transformed or derived values.
- Log any perturbations or deviations from the protocol during the experiment.
- Post-experiment Curation:
- Calculate derived traits (e.g., maximum growth rate μmax, half-saturation constant Ks) using explicitly stated mathematical formulas.
- Package the following together: raw data file, metadata file (in JSON-LD or similar structured format adhering to ISA-Tab standards), a README text file explaining file structure, and the code used for trait calculation.
- Deposit the complete package in a repository such as Zenodo or a domain-specific repository like ESS-DIVE, ensuring all elements are linked.
Protocol: Reproducible Model Parameterization and Execution
Purpose: To create a fully reproducible workflow for parameterizing a biogeochemical model (e.g., a Microbial-ENzyme decomposition model, MEND) with genomic/trait data and executing simulations.
Materials:
- Model source code (e.g., in R, Python, Fortran).
- Parameter dataset (compiled from literature or experiments).
- Environmental forcing data (e.g., temperature, precipitation, substrate input).
- Computational environment (local machine, HPC, or cloud).
Procedure:
- Environment Containerization:
- Create a
DockerfileorSingularity definition filethat specifies the base operating system, installs all required software dependencies (e.g., specific versions of R, Python packages, compilers), and copies the model code into the container. - Build the container image and tag it with a unique identifier.
- Upload the container image to a public registry (e.g., Docker Hub, Singularity Library) or provide the definition file for rebuilding.
- Create a
- Workflow Scripting:
- Write a master script (e.g.,
run_model.shorworkflow.py) that performs these steps in sequence: a. Loads the parameter set from a specified file. b. Loads the environmental driver data. c. Executes the model with the specified parameters and drivers. d. Runs any post-processing analyses (e.g., calculating goodness-of-fit statistics). e. Generates output plots and tables.
- Write a master script (e.g.,
- Version Control and Publication:
- Maintain the model code, parameter files, driver data, workflow scripts, and container definition file in a public Git repository (e.g., GitHub, GitLab).
- Use clear commit messages and release tags for specific manuscript submissions or published versions.
- Upon publication, create a final "release" of the repository and archive it with a DOI on Zenodo, linking the code, data, and container.
Visualization of Workflows and Relationships
Diagram Title: G2E Reproducibility Framework Data Flow
Diagram Title: Standardized Data Generation and Curation Protocol
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Tools for Reproducible G2E Research
| Item/Category | Example(s) | Primary Function in G2E Reproducibility |
|---|---|---|
| Containerization Platforms | Docker, Singularity/Apptainer, Podman | Encapsulates the complete software environment (OS, libraries, code) to guarantee identical execution across systems. |
| Workflow Management Systems | Nextflow, Snakemake, Common Workflow Language (CWL) | Defines, executes, and automates multi-step computational workflows (e.g., from sequence analysis to model input), ensuring process transparency. |
| Version Control Systems | Git (hosted on GitHub, GitLab) | Tracks all changes to code, scripts, and text-based parameter files, enabling collaboration and historical recovery of specific model versions. |
| Metadata Standards & Tools | ISA-Tab framework, OMICS standards, Jupyter Notebooks | Provides structured formats and tools to capture experimental and computational metadata, linking data generation to analysis. |
| Persistent Identifier Services | DOI (via Zenodo, Figshare), RRID (for strains), ORCID (for researchers) | Uniquely and permanently identifies digital objects (data, code), biological resources, and researcher contributions, enabling reliable citation. |
| Public Data Repositories | ESS-DIVE (for G2E), SRA, Zenodo, BioModels | Provides long-term, curated storage with access controls and citation tracking for shared data and models. |
| Open-Source Modeling Languages/Frameworks | R/Python (deSolve, SciPy), Stan, Predictive Ecosystem Analyzer (PEcAn) | Provides transparent, community-vetted platforms for model development, parameter estimation, and uncertainty quantification. |
Conclusion
The Genome-to-Ecosystem (G2E) framework provides a transformative, systematic pathway to harness the explosion of microbial genomic data for predictive modeling in biogeochemistry and biomedicine. By moving from foundational concepts through methodological implementation to rigorous validation, this approach addresses the critical scale mismatch between genes and ecosystem or host phenotypes. Key takeaways include the necessity of a trait-based perspective, the importance of robust mathematical integration of omics data, and the demonstrable improvement in predictive power over traditional models. For biomedical and clinical research, this framework offers a powerful tool to mechanistically model host-microbiome-drug interactions, predict patient-specific metabolic outcomes, and design targeted microbiome-based interventions. Future directions must focus on developing standardized trait databases, improving the mechanistic link between genetic potential and expressed function under dynamic conditions, and creating user-friendly computational platforms to democratize access for the broader research community. The successful adoption of G2E principles promises to usher in a new era of precision in both environmental forecasting and personalized medicine.