Unlocking Microbial Heterogeneity: A Comprehensive Guide to Bacterial Single-Cell RNA-Seq Using 10X Genomics Chromium
This article provides a targeted guide for researchers and drug development professionals exploring bacterial single-cell transcriptomics.
Unlocking Microbial Heterogeneity: A Comprehensive Guide to Bacterial Single-Cell RNA-Seq Using 10X Genomics Chromium
Abstract
This article provides a targeted guide for researchers and drug development professionals exploring bacterial single-cell transcriptomics. We cover foundational principles, from defining bacterial scRNA-seq and its unique challenges to the role of the 10X Chromium platform. A detailed methodological walkthrough includes cell preparation, library construction, and data analysis pipelines. The guide addresses common troubleshooting scenarios and optimization strategies for prokaryotic samples. Finally, we present validation frameworks and comparative analyses with bulk RNA-seq and other platforms, empowering users to design robust studies of bacterial populations at unprecedented resolution.
Why Bacterial Single-Cell RNA-Seq? Overcoming Prokaryotic Challenges with 10X Chromium Technology
Bacterial scRNA-seq represents a paradigm shift from traditional bulk RNA-seq, which averages gene expression across millions of cells, thereby masking cellular heterogeneity. Within the framework of a thesis utilizing the 10X Genomics Chromium platform, this technology enables the dissection of transcriptional states in individual bacterial cells, revealing subpopulations, rare persister cells, and dynamic responses to stressors or drugs with unprecedented resolution.
Application Notes
Bacterial scRNA-seq presents unique challenges, including the need to lyse robust cell walls, capture small and non-polyadenylated mRNAs, and manage high ribosomal RNA content. Successful application enables:
- Antibiotic Resistance & Persistence: Identification of rare, transient subpopulations that survive antibiotic treatment.
- Host-Pathogen Interactions: Profiling of bacterial transcriptional responses within individual host cells.
- Metabolic Heterogeneity: Mapping of division of labor and metabolic specialization in microbial communities.
- Biotechnological Optimization: Screening for high-producing single cells in industrial fermentation.
Table 1: Comparison of Bulk vs. Single-Cell RNA-seq for Bacterial Studies
| Feature | Bulk RNA-seq | Single-Cell RNA-seq (10X Chromium) |
|---|---|---|
| Resolution | Population average | Individual cell |
| Heterogeneity Detection | No | Yes |
| Rare Cell Identification | Not possible | Possible (e.g., persisters) |
| Key Output | Mean expression levels | Expression matrix per cell |
| Primary Challenge | Deconvoluting mixed signals | Technical noise, data sparsity |
| Typical Cells Profiled | 10^6 - 10^7 | 10^3 - 10^4 |
Table 2: Key Metrics from Recent Bacterial scRNA-seq Studies
| Organism Studied | Cells Recovered | Median Genes/Cell | Key Finding | Reference Year |
|---|---|---|---|---|
| Mycobacterium tuberculosis | ~8,500 | ~500 | Identified a drug-tolerant state with upregulated efflux pumps | 2023 |
| Escherichia coli (stationary phase) | ~15,000 | ~400 | Distinguished subpopulations with divergent metabolic activity | 2024 |
| Salmonella Typhimurium (in macrophages) | ~6,000 | ~300 | Mapped distinct intracellular virulence programs | 2023 |
Protocol: 10X Genomics Chromium Workflow for Gram-Negative Bacteria
Principle: This protocol adapts the 10X Chromium Next GEM technology for bacteria, focusing on cell wall disruption and prokaryotic transcript capture.
I. Sample Preparation & Lysis
- Culture & Fixation: Grow bacterial culture to mid-log phase. Stabilize transcriptomes by adding 1/10 volume of ice-cold 5% Phenol:Ethanol (95%) solution directly to culture for 15 min on ice. Pellet cells.
- Cell Wall Weakening: Resuspend pellet in 1 mL Tris-EDTA-Lysozyme (10 mM Tris-HCl, 1 mM EDTA, 1 mg/mL lysozyme, pH 8.0). Incubate 15 min at 30°C.
- Lysis: Add 1 mL of 10X Genomics Lysis Buffer (supplemented with 40 U/mL SUPERase•In RNase Inhibitor). Vortex thoroughly. Incubate on ice for 5 min. Pellet debris (12,000g, 2 min, 4°C). Transfer supernatant (containing RNA) to a new tube.
- RNA Isolation & DNase Treatment: Purify total RNA using a column-based kit with on-column DNase I digestion. Elute in 30 μL nuclease-free water.
- rRNA Depletion: Use a commercial kit (e.g., MICROBExpress, Ribo-Zero Plus) designed for Gram-negative bacteria to deplete 16S and 23S rRNA. Follow kit instructions.
II. 10X Library Construction (Modified)
- Priming: Use random hexamers instead of oligo-dT for reverse transcription. Prepare the Master Mix as per the 10X Single Cell 3' Reagent Kits v3.1 protocol, but replace the RT Primer with 2.5 μM final concentration of random hexamers.
- GEM Generation & Barcoding: Proceed with loading the RNA and Master Mix onto the Chromium Chip B for GEM (Gel Bead-in-emulsion) generation. Within each GEM, reverse transcription occurs, adding a cell-specific barcode and unique molecular identifier (UMI) to each cDNA molecule.
- Post GEM-RT Cleanup & Amplification: Break emulsions, purify cDNA with DynaBeads, and amplify via PCR (12 cycles).
- Library Construction: Fragment, A-tail, and ligate sample index adapters to the amplified cDNA as per the standard protocol. Perform final library amplification (12 cycles).
- QC & Sequencing: Assess library size (~450-550 bp) on a Bioanalyzer. Sequence on an Illumina platform aiming for ≥50,000 reads per cell. Use 28 cycles for Read 1 (barcode/UMI), 10 cycles for i7 index, and 90 cycles for Read 2 (transcript).
Workflow Diagram
Title: Bacterial 10X scRNA-seq Workflow
Analysis & Pathway Visualization
A primary application is elucidating antibiotic stress response pathways at single-cell resolution. A simplified signaling and response network for a beta-lactam antibiotic is shown below.
Title: Bacterial Single-Cell Antibiotic Response
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Reagents for Bacterial 10X scRNA-seq
| Item | Function | Example/Note |
|---|---|---|
| Phenol:Ethanol Fixative | Rapid transcriptome stabilization | 5% phenol in 95% ethanol, ice-cold |
| Lysozyme | Weakens peptidoglycan layer for lysis | Critical for Gram-positive bacteria |
| 10X Lysis Buffer | Complete cell disruption & RNA protection | From 10X kits, supplemented with RNase inhibitor |
| Prokaryotic rRNA Depletion Kit | Removes abundant 16S/23S rRNA | MICROBExpress, Ribo-Zero Plus |
| Random Hexamer Primers | Initiate cDNA synthesis from bacterial RNA | Replaces oligo-dT in 10X RT mix |
| 10X Chromium Single Cell Kit | Creates GEMs for barcoding | 3' Gene Expression v3.1 or later |
| Chromium Chip B | Microfluidic device for partitioning cells | |
| SPRIselect Beads | Size selection and cleanup of cDNA libraries | |
| Cell Ranger Pipeline | Demultiplexing, alignment, counting | Must use a modified prokaryotic reference |
In the context of advancing single-cell RNA sequencing (scRNA-seq) for bacterial research using the 10X Genomics Chromium platform, a fundamental challenge arises: the standard chemistry relies on poly-A tail capture for mRNA enrichment. Bacterial transcripts largely lack polyadenylated tails, rendering the default workflow ineffective. This application note details the adapted methodologies and solutions for capturing and sequencing bacterial transcripts at single-cell resolution, enabling host-pathogen interactions, antibiotic resistance studies, and microbial ecology research.
Key Methodological Adaptations
To overcome the poly-A challenge, researchers have developed and optimized several strategies centered on custom probe design and tailored library preparation.
Probe-Based Capture (RTL-P Method)
This method involves designing custom oligonucleotide probes to capture specific bacterial transcripts.
Protocol: RTL-P (Reverse Transcription with Ligation-mediated Probe Capture)
- Step 1 – Probe Design: Design biotinylated DNA oligonucleotide probes (80-120 nt) targeting the transcriptome of interest. Probes should tile across target genes with appropriate spacing.
- Step 2 – Cell Lysis and Hybridization: Single bacterial cells are encapsulated in droplets (10X Chromium). Lysis is performed, releasing RNA. Custom probes are hybridized to target RNAs in the droplet.
- Step 3 – Capture and Reverse Transcription: Streptavidin beads are introduced to capture biotinylated probe:RNA hybrids. Reverse transcription is performed primed by a universal sequence on the probe.
- Step 4 – cDNA Amplification and Library Construction: Following bead cleanup, cDNA is amplified via PCR. Subsequent library construction follows standard 10X steps (fragmentation, adaptor ligation, indexing).
Poly-A-Independent Whole Transcriptome Amplification
This approach uses random priming instead of poly-dT to initiate cDNA synthesis.
Protocol: Random Primer-Based scRNA-seq for Bacteria
- Step 1 – Cell Encapsulation and Lysis: Single cells are partitioned into 10X droplets followed by immediate chemical lysis.
- Step 2 – Random Primed Reverse Transcription: A reaction mix containing random hexamers (or nonamers) and reverse transcriptase is co-encapsulated. First-strand cDNA synthesis occurs without mRNA selection.
- Step 3 – Template Switching and cDNA Amplification: A template-switching oligo (TSO) is used to append a universal sequence to the 3' end of cDNA, enabling PCR amplification of full-length transcripts.
- Step 4 – Probe-Based Depletion of rRNA: To enrich for mRNA, biotinylated probes targeting conserved bacterial ribosomal RNA (rRNA) sequences are hybridized to the amplified cDNA and removed via streptavidin bead pull-down.
- Step 5 – Library Construction: The rRNA-depleted product is processed through the standard 10X library prep workflow.
Table 1: Comparison of Bacterial scRNA-seq Capture Methods
| Method | Principle | Key Advantage | Key Limitation | Approximate Capture Efficiency* | Primary Application |
|---|---|---|---|---|---|
| RTL-P | Sequence-specific probe hybridization | High specificity for target transcripts; low background | Requires prior genomic knowledge; not discovery-based | 60-75% | Targeted expression profiling (e.g., virulence genes) |
| Random Priming + rRNA Depletion | Whole-transcriptome random priming | Discovery-based; poly-A independent | High ribosomal RNA background (>90% initial reads) | 20-40% (post-depletion) | Exploratory studies, unknown transcripts |
| Custom Poly-A Capture | Enriching native/induced polyadenylated RNAs | Uses standard 10X chemistry | Only captures naturally polyadenylated transcripts (<5% in most bacteria) | <10% | Studies on RNA processing/polyadenylation |
*Capture Efficiency: Estimated percentage of targeted mRNA molecules successfully converted into sequenceable library fragments.
Table 2: Typical Sequencing Metrics for Bacterial scRNA-seq (Chromium Next GEM)
| Metric | RTL-P Method | Random Primer + Depletion |
|---|---|---|
| Reads per Cell | 50,000 - 100,000 | 100,000 - 200,000 |
| Genes Detected per Cell | 100 - 500 (targeted) | 500 - 2,000 (whole transcriptome) |
| rRNA Percentage (final library) | <5% | 10-30% |
| Recommended Sequencing Depth | ~20,000 reads/cell | ~50,000 reads/cell |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Bacterial scRNA-seq
| Item | Function | Example/Note |
|---|---|---|
| 10X Chromium Controller & Kits | Microfluidic partitioning and core reagent delivery. | 10X Genomics Chromium Next GEM Chip. Use Chromium Next GEM Single Cell 3' Kit v3.1 as base. |
| Custom Biotinylated Capture Probes | Hybridize to and tag target bacterial mRNAs for capture. | Designed via IDT or Twist Bioscience. Biotin at 3' end. Pool complexity: ~10,000 probes. |
| Streptavidin Magnetic Beads | Capture probe:RNA complexes. | MyOne Streptavidin C1 Dynabeads. |
| Random Hexamer/Nonamer Primers | Initiate cDNA synthesis independent of poly-A tail. | Included in some NEB or Takara reverse transcription kits. |
| Template Switching Oligo (TSO) | Enables full-length cDNA amplification after random priming. | Use the sequence compatible with your 10X kit (e.g., from SMARTER kits). |
| Biotinylated rRNA Depletion Probes | Remove abundant ribosomal RNA sequences from libraries. | Designed against 16S and 23S rRNA of target species (e.g., Hyb-specific probes). |
| RNase Inhibitor | Protect bacterial mRNA during lysis and RT. | Use a potent inhibitor like Recombinant RNase Inhibitor. |
| Lysozyme/Alternative Lysis Buffer | Efficiently break down bacterial cell walls within droplets. | Optimize concentration for specific species (e.g., S. aureus vs E. coli). |
Visualized Workflows
Diagram Title: Targeted Capture via RTL-P Probes
Diagram Title: Whole Transcriptome Random Priming Workflow
Diagram Title: Bacterial scRNA-seq Core Challenge
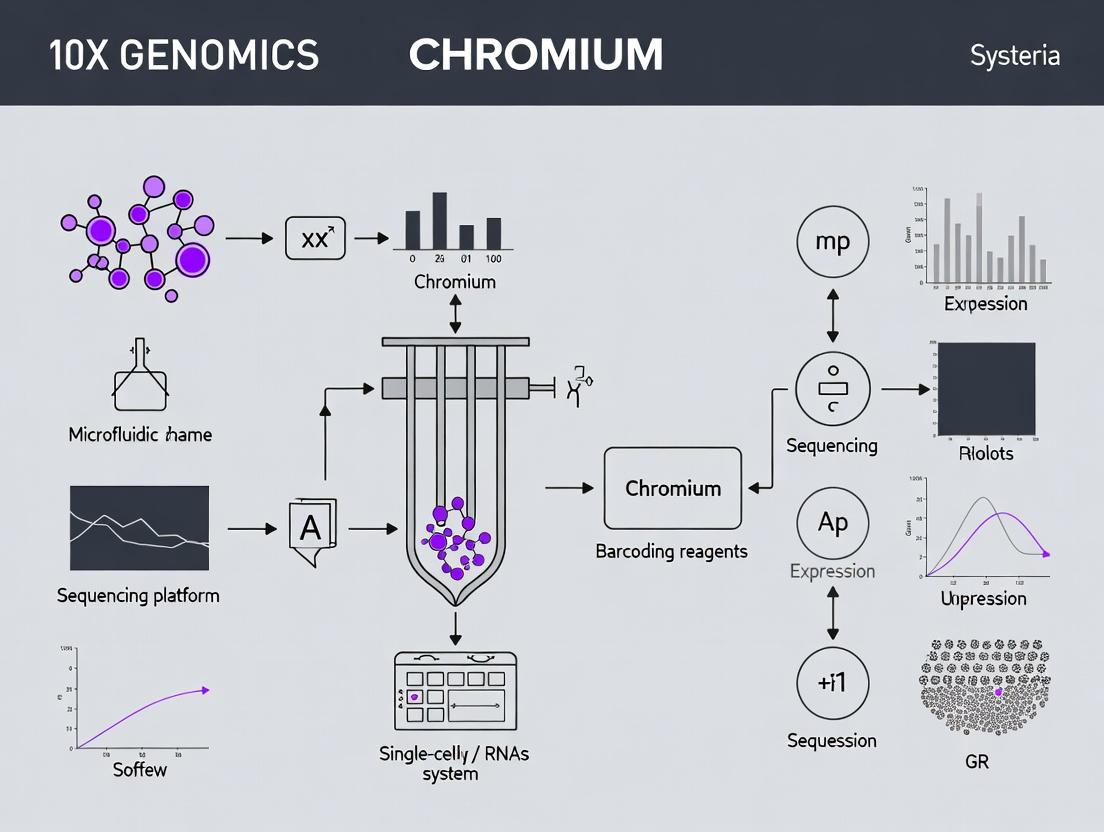
This protocol details the use of the 10X Genomics Chromium Platform, specifically its Gel Bead-in-Emulsion (GEM) technology, for generating single-cell gene expression libraries from bacterial communities. Within the broader thesis on applying 10X Genomics to bacterial single-cell RNA sequencing (scRNA-seq), this technology enables the high-throughput, parallel analysis of transcriptomes from thousands of individual bacterial cells, overcoming challenges related to cell wall lysis and low RNA content. This is critical for research in microbial ecology, host-pathogen interactions, antibiotic resistance heterogeneity, and drug development targeting persistent bacterial subpopulations.
Core Principle of GEM Technology
The fundamental innovation is the Gel Bead-in-Emulsion (GEM). Each GEM is a nanoliter-scale aqueous droplet formed within an oil-surfactant mixture in a microfluidic "Chip" channel. Each droplet functions as an isolated reaction chamber containing:
- A single gel bead with barcoded oligonucleotides.
- A single cell (or partition).
- Enzymes and reagents for Reverse Transcription (RT).
The gel bead dissolves, releasing uniquely barcoded oligonucleotides that tag all cDNA derived from that single cell. This allows pooled sequencing of thousands of cells while retaining single-cell resolution through the unique barcode.
Diagram: GEM Formation and Barcoding Principle
Key Reagent Solutions and Materials
Table 1: The Scientist's Toolkit - Essential Reagents for 10X Bacterial scRNA-seq
| Reagent/Material | Function in Protocol | Key Consideration for Bacteria |
|---|---|---|
| Chromium Next GEM Chip | Microfluidic device to generate GEMs with precise volume control. | Use appropriate chip type (e.g., Chip B) for targeted cell recovery. |
| Barcoded Gel Beads | Contains unique oligos with: 16bp 10X Barcode, 10bp UMI, 30bp Poly(dT) / Gene-Specific primer. | For bacteria (no poly-A tails), custom beads with gene-specific primers (e.g., 16S rRNA primer) are essential. |
| Partitioning Oil & Reagent Kit | Forms stable, uniform emulsions (GEMs) and contains RT master mix. | Must be compatible with downstream bacterial cell lysis chemistry (e.g., enzymatic/chemical). |
| Chromium Controller | Instrument that automates the microfluidic partitioning of cells, beads, and reagents into GEMs. | Standardized run ensures consistent GEM recovery. |
| Cell Lysis Solution | Lyses bacterial cell wall post-GEM formation to release RNA. | Often requires a customized, harsh lysis cocktail (e.g., lysozyme + proteinase K) integrated into the RT mix. |
| Reverse Transcriptase Master Mix | Performs reverse transcription inside each GEM. | Must be robust and efficient for bacterial mRNA templates. |
| Silane Magnetic Beads | For post-GEM cleanup and cDNA purification. | Standard for SPRIselect cleanups. |
| Library Construction Kit | Amplifies barcoded cDNA and adds sample indices and adapters for sequencing. | Follow 10X Genomics protocol for 5' gene expression. |
Detailed Experimental Protocol for Bacterial Applications
Protocol 4.1: GEM Generation and Barcoded cDNA Synthesis
Objective: To partition single bacterial cells into GEMs and generate barcoded, full-length cDNA.
Materials: Chromium Controller, Chip B, 10X Barcoded Gel Beads (custom primer), Partitioning Oil & Reagent Kit, custom RT/Lysis Master Mix, prepared bacterial cell suspension.
Procedure:
- Cell Preparation: Harvest and wash bacterial culture. Resuspend in an appropriate, nuclease-free buffer. Critical: Optimize cell density to maximize single-cell GEMs. Target cell recovery of 1,000-10,000 cells.
- Quantitative Guidance: Use a live/dead stain and cell counter. Aim for a final loading concentration of 700-1200 cells/µL to achieve a multiplet rate <10%.
- Master Mix Preparation: On ice, prepare the RT/Lysis Master Mix as per kit instructions, supplemented with optimized concentrations of bacterial cell wall lytic enzymes.
- Chip Loading: Load the following into the designated wells of a Chromium Chip B:
- Well 1: Prepared cell suspension (70 µL).
- Well 2: Custom barcoded gel beads (40 µL).
- Well 3: RT/Lysis Master Mix (70 µL).
- Well 4: Partitioning Oil (250 µL).
- Run Chromium Controller: Place chip in the Controller and run the "Single Cell 3' v3.1" program (or appropriate custom program). This generates ~80,000 GEMs.
- Reverse Transcription & GEM Incubation: Collect the GEM emulsion (approx. 100 µL) into a PCR tube. Incubate in a thermal cycler:
- 53°C for 45 min (RT).
- 85°C for 5 min (enzyme inactivation).
- Hold at 4°C.
- GEM Breakage and cDNA Cleanup:
- Break the emulsion by adding Recovery Agent, mix, and incubate at room temperature.
- Add Dynabeads Silane magnetic beads to isolate barcoded cDNA.
- Perform two 80% ethanol washes.
- Elute cDNA in 40 µL of Buffer EB.
Protocol 4.2: Library Construction and Sequencing
Objective: To amplify barcoded cDNA and construct Illumina-compatible sequencing libraries.
Materials: SPRIselect Reagent Kit, Sample Index Plate, PCR Enzymes, Library Construction Reagents.
Procedure:
- cDNA Amplification: Perform PCR on the purified cDNA to generate sufficient mass for library construction. Determine cycle number using a qPCR side reaction or based on estimated cell count.
- Typical Cycles: 10-14 cycles.
- cDNA Size Selection: Clean amplified cDNA with SPRIselect beads. Perform a double-sided size selection (e.g., 0.6x / 0.8x ratios) to remove short fragments and primer dimers.
- Library Construction (End Repair, A-tailing, Adapter Ligation): Follow the 10X Genomics protocol to fragment the cDNA, add adapters, and ligate sample index-specific dual indexes via a second PCR (10-14 cycles).
- Library QC and Sequencing:
- Quantify using a fluorescence-based assay (e.g., Qubit).
- Assess fragment size distribution (e.g., Bioanalyzer/TapeStation).
- Pool libraries and sequence on an Illumina platform.
Table 2: Recommended Sequencing Parameters
| Parameter | Recommended Specification | Reason |
|---|---|---|
| Read 1 | 28 cycles | Sequences the 16bp 10X Barcode and 10bp UMI. |
| i7 Index | 10 cycles | Sample index. |
| i5 Index | 10 cycles | Sample index. |
| Read 2 | 90-150 cycles | Sequences the cDNA insert (bacterial transcript). |
| Read Depth | 50,000 - 100,000 reads/cell | Higher depth may be needed for bacterial transcriptomes. |
Data Output and Analysis Workflow
Diagram: From GEMs to Single-Cell Data
Table 3: Key Quantitative Output Metrics for QC
| Metric | Typical Target Range (Mammalian) | Note for Bacterial Adaptation |
|---|---|---|
| Number of Cells Recovered | User-defined (e.g., 10,000) | Lower due to size/lysis; aim for 1,000-5,000 high-quality cells. |
| Median Genes per Cell | 1,000 - 5,000 | Significantly lower for bacteria (tens to hundreds). Requires adjusted thresholds. |
| Median UMI Counts per Cell | 10,000 - 50,000 | Lower for bacteria. Indicator of lysis & RT efficiency. |
| GEM Saturation | >50% | Measures sequencing depth for transcript detection. |
| Fraction of Reads in Cells | >70% | Lower values may indicate high ambient RNA or cell debris. |
Application Notes
Within the broader thesis of applying 10X Genomics Chromium technology to bacterial single-cell RNA sequencing (scRNA-seq), three transformative applications emerge. These address long-standing challenges in microbiology by enabling the dissection of phenotypic heterogeneity in bacterial populations at unprecedented resolution.
1. Studying Antibiotic Persistence: Traditional bulk RNA-seq averages the response of a bacterial population to antibiotic stress, masking rare, transient subpopulations known as persisters. Chromium-based 3' scRNA-seq allows for the isolation and transcriptional profiling of individual bacterial cells, identifying distinct persister states characterized by upregulated stress-response pathways (e.g., SOS, toxin-antitoxin systems) and downregulated metabolic activity. This reveals the regulatory networks driving tolerance, moving beyond the stochastic model to defined cell states.
2. Elucidating Host-Pathogen Interactions: During infection, both host and pathogen undergo dynamic, heterogeneous changes. Dual RNA-seq at single-cell resolution is now possible. By combining the 10X Chromium Fixed RNA Profiling Kit (for host eukaryotic cells) with custom workflows for bacterial RNA capture, one can simultaneously profile the transcriptional states of infected host cells (e.g., macrophage polarization states) and the intracellular bacterial pathogens they contain. This reveals coordinated and antagonistic gene programs, identifying key virulence strategies and host defense mechanisms at the level of individual infection events.
3. Decoding Microbial Community Heterogeneity: In complex consortia like the gut microbiome, function is dictated by the combined activity of myriad species and strains. Single-cell partitioning followed by cDNA amplification and metagenomic analysis (e.g., using the Chromium Genome or Custom assays) enables the linking of taxonomic identity (via conserved genomic regions) to functional potential (via RNA expression) for thousands of individual microbes in parallel. This resolves strain-level functional diversity, metabolic cross-feeding interactions, and niche specialization without the need for cultivation.
Quantitative Data Summary: Key Metrics for Bacterial scRNA-seq on 10X Chromium
| Application | Typical Cell Recovery | Recommended Sequencing Depth per Cell | Key Output Metric | Representative Reference |
|---|---|---|---|---|
| Antibiotic Persistence | 5,000 - 10,000 cells | 20,000 - 50,000 reads | % cells in distinct persister state cluster; differential expression of stress genes. | Blattman et al., 2020 (Nat. Microbiol.) |
| Host-Pathogen (Dual) | 1,000 - 5,000 host cells | 50,000+ reads (host) | # of pathogen transcripts per infected host cell; correlated host-pathogen gene modules. | Dieterich et al., 2023 (Cell Host & Microbe) |
| Microbial Communities | 10,000+ microbial cells | 10,000 - 30,000 reads | Species/Strain abundance linked to functional gene expression profiles. | Woyke et al., 2021 (Science Advances) |
Experimental Protocols
Protocol 1: Single-Cell RNA-seq of Antibiotic-Treated Escherichia coli for Persister Analysis
Objective: To identify and transcriptionally characterize bacterial persister cells following antibiotic challenge.
Materials: See "The Scientist's Toolkit" below.
Method:
- Culture & Treatment: Grow E. coli to mid-log phase (OD600 ~0.3-0.4). Treat a sample with a lethal dose of a bactericidal antibiotic (e.g., 10x MIC of ciprofloxacin or ampicillin) for 2-4 hours. Include an untreated control.
- Persister Isolation & Fixation: Wash treated culture 2x with PBS. Resuspend in 4% paraformaldehyde (PFA) in PBS and fix for 1 hour at room temperature. Quench with 0.1M glycine. Note: Fixation is critical for safety and RNA stability.
- Permeabilization & Hybridization (Custom): Pellet cells, resuspend in 70% ice-cold ethanol, and store at -20°C for up to 1 week. Rehydrate with wash buffer. Apply a custom panel of gene-specific, DNA-barcoded probes targeting key bacterial mRNAs (e.g., recA, relA, hipA, ribosomal genes).
- 10X Library Preparation: Follow the 10X Genomics Chromium Fixed RNA Profiling Protocol (Document CG000583) from the cell suspension step, using the probe-hybridized, fixed bacterial cells as input. Use a custom probe set file.
- Sequencing & Analysis: Sequence on an Illumina platform (e.g., NovaSeq) to achieve ~30,000 reads/cell. Process using Cell Ranger with a custom probe reference. Cluster cells using Seurat or Scanpy. Persisters will appear as distinct clusters with unique transcriptional signatures.
Protocol 2: Dual Host-Pathogen Single-Cell RNA-seq from Infected Macrophages
Objective: To capture paired transcriptional profiles from individual infected mammalian host cells and their intracellular bacterial cargo.
Materials: See "The Scientist's Toolkit" below.
Method:
- Infection & Fixation: Infect a monolayer of murine bone-marrow-derived macrophages (BMDMs) with Salmonella enterica (MOI ~5) for 6-12 hours. Gently wash away extracellular bacteria. Fix cells with 4% PFA for 30 min, quench with glycine, and scrape.
- Probe Hybridization (Dual): Permeabilize fixed cells. Split sample into two aliquots:
- Aliquot A (Host): Apply the Fixed RNA Profiling Probe Barcode library for mouse transcriptome.
- Aliquot B (Pathogen): Apply a custom probe panel for key Salmonella virulence (sipB, sifA, ssaG) and stress genes.
- Cell Partitioning & Library Prep: Pool the two aliquots. Load onto the Chromium Controller using the Fixed RNA Profiling Chip. The Gel Beads contain two distinct capture sequences: one for host probe barcodes and one for custom bacterial probe barcodes. Proceed with extension, ligation, and library construction per the standard protocol.
- Sequencing & Demultiplexing: Sequence deeply (>50K reads/cell). Use Cell Ranger with a combined reference (mouse genome + custom probe set) to generate a unified feature-barcode matrix.
- Data Analysis: Identify infected host cells by the presence of bacterial probe UMIs. Perform integrated analysis to correlate host immune pathways (e.g., inflammasome genes) with bacterial virulence programs.
Mandatory Visualization
Title: Workflow for Single-Cell Analysis of Antibiotic Persistence
Title: Host-Pathogen Interaction Pathways Revealed by Dual scRNA-seq
Title: Microbial Community Analysis by Linked Single-Cell RNA/DNA Sequencing
The Scientist's Toolkit: Essential Research Reagents & Materials
| Item | Function/Description | Key Supplier/Example |
|---|---|---|
| 10X Chromium Controller & Chip K | Microfluidic platform to partition single cells into Gel Bead-in-Emulsions (GEMs). | 10X Genomics (PN-1000154) |
| Chromium Fixed RNA Profiling Kit | Core kit for capturing probe-barcoded RNA from fixed cells. Contains Gel Beads, reagents, and buffers. | 10X Genomics (PN-1000490) |
| Custom Probe Panels (xGen) | Designer DNA oligonucleotide probes targeting specific bacterial mRNA transcripts for capture. | Integrated DNA Technologies (IDT) |
| Paraformaldehyde (4%, ampulated) | For immediate, consistent cross-linking of cells to preserve RNA state and inactivate pathogens. | Thermo Fisher Scientific (PN-043368.9M) |
| Glycine (1M solution) | Quenching agent to stop the fixation reaction by reacting with excess PFA. | Sigma-Aldrich (PN-G7126) |
| Protease Inhibitor Cocktail | Added during cell lysis to prevent degradation of proteins/DNA in complex community samples. | Roche (cOmplete, PN-04693116001) |
| RNase Inhibitor | Critical for all steps post-fixation to protect bacterial mRNA, which is less polyadenylated. | Lucigen (RNASecure, PN-21200) |
| Magnetic Stand (for 0.2mL tubes) | For clean purification of cDNA and libraries using SPRIselect beads. | Thermo Fisher Scientific (DynaMag-96) |
| SPRIselect Beads | Size-selective magnetic beads for cleanup and size selection of cDNA and final libraries. | Beckman Coulter (PN-B23318) |
| Bioanalyzer High Sensitivity DNA Kit | Quality control of final libraries pre-sequencing to assess fragment size distribution. | Agilent Technologies (PN-5067-4626) |
1. Introduction and Thesis Context Within the broader thesis on applying the 10X Genomics Chromium platform to bacterial single-cell RNA-seq research, the foundational stage is critical. Success depends on meticulous preparation, recognizing that bacterial studies face unique challenges such as cell wall lysis, lack of polyadenylated tails, and high ribosomal RNA content. This document outlines the essential prerequisites for designing a robust bacterial scRNA-seq study.
2. Essential Lab Setup and Safety Considerations A dedicated pre-PCR workspace is mandatory to prevent contamination. The setup must be organized into distinct zones:
- Zone 1 (Clean Reagent Prep): For master mix preparation, away from all nucleic acid sources.
- Zone 2 (Sample Processing & Cell Lysis): Equipped with a biosafety cabinet for handling bacterial cultures. Includes a microcentrifuge, vortexer, and temperature-controlled incubator/thermal cycler for enzymatic lysis steps.
- Zone 3 (Post-Lysis & Library Construction): Contains equipment for cDNA amplification and library building, including a verified PCR machine, magnetic separator, and qPCR system for QC.
All work with live bacteria must adhere to appropriate Biosafety Level (BSL) guidelines. RNase-free consumables and dedicated pipettes are required throughout.
3. Sample Types, Selection, and Preparation Sample integrity is paramount. Key considerations are summarized in Table 1.
Table 1: Bacterial Sample Types and Preparation Requirements
| Sample Type | Key Characteristics | Critical Preparation Notes | Compatibility with 10X Protocol |
|---|---|---|---|
| Planktonic Cells | Homogeneous suspension, standard lab condition. | Requires precise OD600 measurement; viability >90% recommended. Filter through cell strainer (e.g., 35µm) to remove clumps. | High. Standard cell suspension protocols apply after lysis. |
| Biofilms | Structured, heterogeneous, matrix-encased communities. | Requires robust mechanical (sonication, bead beating) and/or enzymatic (Dispase, DNase I) disruption. Post-disruption, intensive washing to remove debris. | Moderate. Debris and extracellular DNA can clog microfluidics. |
| Tissue-associated/Intracellular Bacteria | Bacteria extracted from host cells or tissue. | Host cell lysis followed by bacterial enrichment (differential centrifugation, gradient separation). Must deplete host nucleic acid background. | Low-Moderate. Purity is the major challenge; host contamination can dominate sequencing. |
| Environmental Isolates | Unknown or variable cell wall composition. | Lysis conditions must be empirically tested (enzymatic vs. chemical). Viability assessment may be challenging. | Variable. Highly dependent on successful lysis and obtaining single-cell suspensions. |
4. Initial Planning and Experimental Design A successful plan addresses the following steps, as visualized in the workflow diagram.
Diagram 1: Initial Planning Workflow for Bacterial scRNA-seq
4.1 Detailed Protocol: Optimization of Bacterial Cell Lysis for Single-Cell Capture Objective: To achieve complete lysis while maintaining RNA integrity and compatibility with 10X microfluidics. Reagents: Tris-EDTA Buffer, Lysozyme (10-100 mg/mL), Mutanolysin (for Gram-positives), Proteinase K, RNase Inhibitor. Procedure:
- Harvest & Wash: Pellet 1x10^8 bacterial cells. Wash 2x in ice-cold PBS or suitable buffer.
- Enzymatic Lysis: Resuspend pellet in 100µL Tris-EDTA buffer with RNase Inhibitor. Add lysozyme (final 10 mg/mL). For Gram-positives, add mutanolysin (final 0.1 U/µL).
- Incubate: 37°C for 15-30 minutes. Monitor lysis microscopically or by viscosity.
- Protein Digestion (Optional): Add Proteinase K (final 0.5 mg/mL). Incubate at 55°C for 10 min.
- Enzyme Inactivation: Place on ice. Optionally, heat-inactivate at 70°C for 10 min.
- Debris Removal: Centrifuge at max speed, 4°C for 5 min. Transfer supernatant (containing RNA) to a fresh tube.
- QC: Check RNA integrity (RIN >7.5 ideal) via Bioanalyzer/TapeStation and concentration via Qubit RNA HS Assay.
4.2 Detailed Protocol: rRNA Depletion via Probe-based Hybridization Objective: To deplete abundant bacterial rRNA prior to cDNA synthesis. Reagents: Ribo-zero rRNA Removal Kit (Bacteria), RNase H, RNase-free DNase I, Magnetic Beads (SPRI). Procedure:
- Denature RNA: Mix up to 5µg of total bacterial RNA with hybridization buffer. Heat at 70°C for 2 min, then immediately place on ice.
- Hybridize: Add sequence-specific rRNA depletion probes. Incubate at 70°C for 5 min, then 37°C for 15 min.
- Remove rRNA-Probe Hybrids: Add magnetic beads prepared according to kit. Incubate at room temperature.
- Capture: Place on magnet. Transfer supernatant (rRNA-depleted RNA) to new tube.
- Cleanup: Perform two rounds of SPRI bead cleanup. Elute in nuclease-free water.
- QC: Assess depletion efficiency via Bioanalyzer (prokaryote total RNA assay).
5. The Scientist's Toolkit: Key Research Reagent Solutions Table 2: Essential Materials for Bacterial scRNA-seq Sample Prep
| Item | Function/Application | Example Product/Type |
|---|---|---|
| RNase Inhibitor | Protects RNA from degradation during all steps. | Recombinant Ribonuclease Inhibitor |
| Lysozyme | Degrades peptidoglycan layer of bacterial cell walls. | Lyophilized powder, molecular biology grade |
| Mutanolysin | Cleaves peptidoglycan (especially effective on Gram+). | From Streptomyces globisporus |
| Ribo-zero rRNA Removal Kit (Bacteria) | Depletes 5S, 16S, and 23S rRNA via probe hybridization. | Illumina Ribo-Zero Plus |
| Template Switching Oligo (TSO) | Enables template-switching during RT for cDNA amplification. | Required for 10X 5' or 3' v3.1 kits |
| Random Hexamer Primers | Initiate reverse transcription across bacterial transcripts lacking poly-A tails. | Nuclease-free, HPLC purified |
| SPRIselect Magnetic Beads | Size selection and cleanup of cDNA and libraries. | Beckman Coulter SPRIselect |
| Cell Strainer | Removes cell aggregates and debris prior to loading on Chromium Chip. | 35µm nylon mesh, low protein binding |
| Live/Dead Cell Stain | Assesses bacterial cell viability pre-lysis (if using intact cells). | SYTO BC / Propidium Iodide |
| Chromium Next GEM Chip K | Microfluidic device for single-cell partitioning. | 10X Genomics Chip K (for 16 reactions) |
Step-by-Step Protocol: From Bacterial Culture to Single-Cell Library on the 10X Chromium
This application note details the critical first step in bacterial single-cell RNA sequencing (scRNA-seq) workflows optimized for the 10X Genomics Chromium platform. The effective capture of transcriptional states in prokaryotes requires specialized protocols to address their unique cell wall structures, small size, and lack of polyadenylated tails. This content supports a broader thesis on adapting Chromium technology for microbial research, enabling insights into population heterogeneity, antibiotic persistence, and host-pathogen interactions for drug development.
Key Challenges & Considerations
Bacterial scRNA-seq presents distinct challenges: 1) Cell Wall Integrity: Gram-positive and Gram-negative bacteria require different permeabilization strategies. 2) mRNA Capture: Bacterial mRNA lacks poly-A tails, necessitating custom capture probes or polyadenylation treatments. 3) Ribosomal RNA (rRNA) Depletion: >90% of bacterial RNA is rRNA, requiring efficient depletion to enrich mRNA. 4) Cell Size: Small bacterial cells (0.5-2 µm) must be efficiently encapsulated in droplets.
Table 1: Comparison of Fixation and Permeabilization Reagents for Bacterial Cells
| Reagent | Primary Function | Optimal Concentration | Incubation Time (RT) | Target Cell Type | Key Advantage | Key Limitation |
|---|---|---|---|---|---|---|
| Formaldehyde (FA) | Crosslinking fixative | 1-4% (v/v) | 15-30 min | Gram-/+ | Excellent morphology preservation; reversible crosslinks | Over-fixation reduces RNA yield |
| Ethanol | Dehydrating fixative | 70% (v/v) | ≥1 hour, 4°C | Gram-/+ | Simplicity; good for many downstream assays | Can be less effective for some Gram+ species |
| Lysozyme | Peptidoglycan digestion | 1-10 mg/mL | 15-30 min, 30°C | Gram-/+ (Gram+ > Gram-) | Enzymatic, specific cell wall weakening | Activity varies by species/buffer conditions |
| Glycopeptidase | Peptidoglycan digestion | 100 µg/mL | 30 min, 37°C | Gram+ | Highly effective for thick peptidoglycan layer | Expensive; requires precise buffer |
| EDTA + Tris | Membrane destabilization | 10 mM EDTA, 10 mM Tris | 10 min, 4°C | Gram- (Outer membrane) | Chelates Mg2+ to destabilize LPS layer | Ineffective against Gram+ alone |
| Triton X-100 | Non-ionic detergent | 0.1-0.5% (v/v) | 10-15 min, 4°C | Gram-/+ (Post-enzyme) | Mild permeabilization of inner membrane | Can inhibit reverse transcription |
Table 2: Protocol Performance Metrics for Model Organisms
| Bacterial Species | Cell Wall Type | Recommended Harvesting | Fixation Method | Permeabilization Strategy | Median UMIs/Cell* | rRNA% Post-Depletion* |
|---|---|---|---|---|---|---|
| Escherichia coli | Gram-negative | Rapid filtration (0.22µm) | 2% FA, 15 min | 0.2% Triton X-100, 10 min | ~1,200 | 35% |
| Bacillus subtilis | Gram-positive | Centrifugation (5,000 x g) | 70% EtOH, 1hr | 5 mg/mL Lysozyme, 20 min | ~950 | 45% |
| Mycobacterium smegmatis | Mycolic Acid | Gentle centrifugation | 4% FA, 20 min | Glycopeptidase + 0.1% SDS | ~800 | 55% |
| Pseudomonas aeruginosa | Gram-negative | Filter + ice-cold PBS | 1% FA + 0.05% Glutaraldehyde, 10 min | 10mM EDTA-Tris + 0.1% Triton | ~1,500 | 40% |
*Example metrics from adapted 10X workflows; actual results vary by sample prep and probe panel.
Detailed Protocols
Protocol A: Harvesting and Fixation for Gram-Negative Bacteria (e.g.,E. coli)
Goal: Capture cells in mid-log phase and fix transcriptional state with minimal perturbation. Materials: Ice-cold 1X PBS (RNase-free), 16% Formaldehyde (methanol-free, RNase-free), 2.5M Glycine (RNase-free), 0.22µm filter unit or centrifuge.
- Culture & Harvest: Grow cells to mid-log phase (OD600 ~0.3-0.5). Immediately pour culture over a 0.22µm sterile filter membrane under gentle vacuum. Alternatively, rapidly chill culture on ice and centrifuge at 4,000 x g for 5 min at 4°C.
- Wash: Resuspend cell pellet in 10 mL ice-cold 1X PBS. Repeat centrifugation. Final pellet should be ~1x10^8 cells.
- Fixation: Resuspend pellet in 1 mL PBS. Add 1 mL of 4% Formaldehyde (diluted from 16% stock in PBS) for a final concentration of 2%. Incubate at room temperature for 15 minutes with gentle rotation.
- Quenching: Add 125 µL of 2.5M Glycine (final ~250 mM) to quench fixation. Incubate 5 min at RT.
- Wash & Store: Pellet cells at 4,000 x g for 5 min. Wash 2x with 1 mL ice-cold PBS. Resuspend in 1 mL PBS + 0.1% BSA. Store at 4°C for up to 24 hours before permeabilization, or pellet and store at -80°C in PBS/BSA with 10% DMSO.
Protocol B: Enzymatic Permeabilization for Gram-Positive Bacteria (e.g.,B. subtilis)
Goal: Weaken thick peptidoglycan layer to allow access to cytoplasmic RNA. Materials: TE Buffer (10 mM Tris-Cl, 1 mM EDTA, pH 8.0), Lysozyme (RNase-free), RNase Inhibitor, 0.1% Triton X-100.
- Post-Fixation Processing: Begin with ethanol-fixed cells (from 70% EtOH, 1hr at 4°C). Pellet 1x10^8 cells at 5,000 x g for 5 min. Remove ethanol completely.
- Enzymatic Treatment: Resuspend pellet in 100 µL TE Buffer containing 5 mg/mL Lysozyme and 1 U/µL RNase Inhibitor. Incubate at 30°C for 20 minutes. Gently mix every 5 min.
- Detergent Permeabilization: Add 900 µL of ice-cold PBS containing 0.1% Triton X-100. Mix gently and incubate on ice for 10 minutes.
- Wash & Resuspend: Pellet cells at 4,000 x g for 5 min at 4°C. Wash twice with 1 mL of 1X PBS + 0.1% BSA + RNase Inhibitor.
- QC & Proceed: Check cell integrity under microscope. Resuspend in appropriate resuspension buffer (e.g., 10X Genomics Diluent C) at ~1,000 cells/µL for targeting cell recovery.
Protocol C: Integrated Workflow for 10X Chromium Library Prep
Goal: Generate barcoded cDNA from fixed/permeabilized bacterial cells for sequencing. Note: This protocol assumes the use of a custom bacterial probe panel (e.g., designed with the 10X Feature Barcode technology) to capture mRNA.
- Cell Suspension QC: Adjust fixed/permeabilized cell concentration to 1,000-1,500 cells/µL in Diluent C. Pass through a 35µm cell strainer cap.
- Chromium Chip Loading: Load the cell suspension, gel beads (from a 10X Single Cell 3' v3.1 kit), and partitioning oil into a Chromium Chip B according to manufacturer's instructions. Target recovery: 5,000-10,000 cells.
- In-Droplet Processing (GEMs): Post-partitioning, perform on-bead lysis (if permeabilization is partial), reverse transcription using gene-specific primers from the custom panel, and cDNA amplification via PCR.
- rRNA Depletion & Library Prep: Use post-cDNA amplification cleanup. Perform rRNA depletion using sequence-specific RNase H digestion or probe-based pull-down. Construct libraries per 10X protocol with sample index PCR.
- Sequencing: Pool libraries and sequence on an Illumina platform. Recommended read depth: ≥50,000 reads/cell.
Visualizations
Title: Bacterial scRNA-seq Workflow for 10X Genomics
Title: Permeabilization Strategies by Cell Wall Type
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions
| Item | Function & Rationale | Example Product/Catalog |
|---|---|---|
| Methanol-Free Formaldehyde (16%) | Crosslinking fixative. Preserves RNA-protein complexes while maintaining RNA accessibility for RT. Methanol-free reduces RNA degradation. | Thermo Fisher Scientific, 28906 |
| RNase Inhibitor (Murine or Recombinant) | Essential for all steps post-fixation. Inactivates RNases released during permeabilization to protect bacterial mRNA. | Protector RNase Inhibitor, Sigma-Aldrich, 3335402001 |
| Lysozyme (Molecular Biology Grade) | Hydrolyzes β-1,4-glycosidic bonds in peptidoglycan. Critical for Gram-positive permeabilization. Must be RNase-free. | Sigma-Aldrich, L4919 |
| Triton X-100 Detergent | Non-ionic surfactant. Disrupts lipid bilayers (inner membrane) after cell wall digestion. Used at low concentrations. | Sigma-Aldrich, X100 |
| Glycopeptidase (or Mutanolysin) | Cleaves peptidoglycan specifically between muramic acid and L-alanine. Effective for stubborn Gram-positive species. | Cosmo Bio, GPE-10 |
| 0.1% Diethylpyrocarbonate (DEPC)-treated Water | Used to make all aqueous solutions RNase-free by inactivating RNases. | Thermo Fisher Scientific, 750023 |
| Diluent C (10X Genomics) | Optimized buffer for resuspending fixed cells prior to loading on Chromium. Maintains cell viability and integrity. | 10X Genomics, 2000273 |
| 35µm Cell Strainer Snap Cap | Removes cell clumps and debris prior to loading on Chromium chip to prevent microfluidic clogging. | Flowmi Cell Strainer, Sigma, BAH136800040 |
| Custom Bacterial Probe Panel | Set of gene-specific, biotinylated DNA probes to capture bacterial mRNA (lacking poly-A tails) in the 10X workflow. | Designed via 10X Custom Panel Builder or service providers like IDT. |
Within a broader thesis employing the 10X Genomics Chromium platform for bacterial single-cell RNA sequencing, a critical challenge is the selective capture of informative mRNA. Prokaryotic transcripts lack poly-A tails, and total RNA is dominated (>90%) by ribosomal RNA (rRNA). Custom probe-based hybridization capture is therefore an essential step to either deplete rRNA or enrich for targeted gene panels, enabling cost-effective and sensitive sequencing of meaningful transcripts in single-bacterial-cell applications.
Core Probe Design Strategies
rRNA Depletion Probes
This approach uses biotinylated DNA oligonucleotides complementary to conserved regions of the target organism's 5S, 16S, and 23S rRNA. Hybridization followed by streptavidin bead pull-down removes rRNA from the lysate.
Gene-Specific Panels
Panels of biotinylated probes are designed against a curated set of genes of interest (e.g., virulence factors, antibiotic resistance genes, key metabolic pathways). This positive-enrichment strategy is ideal for focused studies.
Table 1: Comparison of Probe Design Strategies for Bacterial scRNA-seq
| Feature | rRNA Depletion | Gene-Specific Panel |
|---|---|---|
| Primary Goal | Remove abundant non-coding RNA | Encode specific mRNA targets |
| Probe Design Basis | Align to conserved rRNA sequences | Align to specific open reading frames |
| Typical Probe Length | 70-120 nt DNA oligos | 70-120 nt DNA oligos |
| Coverage | Whole transcriptome (after depletion) | Targeted subset (100-5000 genes) |
| Best For | Exploratory/discovery research | Focused hypothesis testing |
| Compatibility with 10X | Integrated post-lysis, pre-RT | Integrated post-lysis, pre-RT |
| Estimated Capture Efficiency | 85-99% rRNA removal | 70-90% on-target rate for panel genes |
Detailed Protocol: Hybridization Capture for rRNA Depletion in 10X Bacterial Workflow
Important Note: This protocol is designed to be inserted after bacterial cell lysis within a single-cell partition (GEM) but before reverse transcription in a modified 10X workflow.
Materials & Reagents
- Lysate from 10X GEMs (containing total RNA from encapsulated bacteria).
- Biotinylated rRNA Depletion Probes (Pool, 100 µM total in TE buffer).
- Hybridization Buffer (2X): 40% Formamide, 12x SSC, 20 mM EDTA, 0.2% Tween-20.
- Streptavidin Magnetic Beads (e.g., MyOne C1).
- Bead Wash Buffer (1X): 10 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.1% Tween-20.
- Nuclease-free Water.
- Magnetic Separation Rack.
- Thermal Cycler.
Procedure
Probe Hybridization:
- Combine in a PCR tube:
- 10 µL GEM lysate (total RNA).
- 2 µL Biotinylated Probe Pool (final ~2 µM each probe).
- 12 µL 2X Hybridization Buffer.
- Mix thoroughly and incubate in a thermal cycler: 95°C for 2 min (denature), then 50°C for 15 min (hybridize).
- Combine in a PCR tube:
Bead Preparation:
- Resuspend Streptavidin beads.
- Transfer 40 µL bead suspension per reaction to a new tube.
- Place on magnetic rack for 1 min. Remove supernatant.
- Wash beads twice with 100 µL 1X Bead Wash Buffer. Resuspend in 24 µL 1X Hybridization Buffer.
Capture & Wash:
- Transfer the 24 µL hybridization reaction to the tube with washed beads. Mix gently.
- Incubate at 50°C for 15 min with intermittent mixing to capture probe-rRNA complexes.
- Place tube on magnetic rack for 2 min. Carefully transfer supernatant (containing depleted RNA) to a new tube.
- Optional: Add 20 µL 1X Hybridization Buffer to beads, mix, and perform a second magnetic separation. Pool this wash with the first supernatant to maximize yield.
Clean-up & Continuation:
- Purify the pooled supernatant (~44 µL) using a standard RNA Cleanup Kit (e.g., SPRIselect beads at 1.8X ratio).
- Elute RNA in 10 µL nuclease-free water.
- This eluate is now rRNA-depleted RNA ready for the 10X Genomics Reverse Transcription step.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Custom Probe Capture
| Item | Function & Critical Notes |
|---|---|
| Biotinylated DNA Oligo Pool (Custom) | The core reagent; specificity defined by sequence. HPLC-purified. Modified with 3' or 5' Biotin-TEG. |
| MyOne Streptavidin C1 Beads | High binding capacity magnetic beads for efficient pull-down of biotinylated complexes. |
| Formamide (Molecular Biology Grade) | Denaturant in hybridization buffer; lowers required temperature for specific binding. |
| 20X SSC Buffer | Provides ionic strength (salinity) for hybridization buffer; critical for probe kinetics. |
| SPRIselect Beads | For post-capture clean-up and size selection, integrating with 10X workflow steps. |
| 10X Gel Bead-in-emulsion (GEM) Kit | The core single-cell partitioning and barcoding system. Protocol is modified to include capture step. |
| Thermostable RNase Inhibitor | Added to hybridization mix to protect mRNA during elevated temperature steps. |
Workflow & Decision Pathway Diagrams
Diagram 1 Title: Probe Strategy Decision Workflow for Bacterial scRNA-seq
Diagram 2 Title: Custom Probe Depletion Integrated into 10X Protocol
This Application Note details the critical adaptation of the standard 10X Chromium single-cell RNA-seq workflow for prokaryotic systems. Within the broader thesis on utilizing 10X Genomics for bacterial single-cell research, this step addresses the fundamental choice between 3’ Gene Expression and 5’ assays, which is dictated by the lack of polyadenylated tails in bacterial mRNA. Successful bacterial scRNA-seq requires tailored chemistry and protocols to capture native bacterial transcripts.
Core Comparison: 3’ vs. 5’ Chemistry for Bacteria
The standard 10X 3’ Gene Expression kit relies on poly-dT priming to capture eukaryotic mRNA. Bacterial mRNA lacks poly-A tails, necessitating a switch to a 5' assay that uses random priming and template switching for cDNA synthesis.
Table 1: Quantitative & Functional Comparison of 3’ vs. Adapted 5’ Assays for Bacteria
| Feature | Standard 3’ Gene Expression Kit | Adapted 5’ Assay for Bacteria |
|---|---|---|
| Target | Eukaryotic polyadenylated (poly-A+) mRNA | Total bacterial RNA (rRNA-depleted) |
| Priming Chemistry | Poly-dT primers | Random hexamers + Template Switch Oligo (TSO) |
| Compatible 10X Kit | Chromium Next GEM Single Cell 3’ | Chromium Next GEM Single Cell 5’ |
| Key Adaptation | Not applicable for native bacterial RNA | Requires custom Gel Beads with random primers instead of poly-dT. |
| rRNA Handling | Poly-A selection naturally depletes rRNA | Requires external rRNA depletion (e.g., probe-based) prior to loading. |
| Gene Coverage Bias | 3’ biased | More uniform 5’ to 3’ coverage. |
| Typical Cell Throughput | 500 – 10,000 cells | 500 – 10,000 cells |
| Estimated Bacterial Capture Efficiency* | <1% (without poly-A tailing) | 10-45% (post-rRNA depletion) |
| Primary Challenge | Failure to capture most mRNA. | Optimization of rRNA depletion and cell lysis. |
*Efficiency varies based on species, rRNA depletion method, and lysis efficacy.
Detailed Experimental Protocol
Protocol: Bacterial Single-Cell RNA-seq using Adapted 10X 5’ Chemistry
A. Pre-sequencing Sample Preparation (Day 1) Objective: Generate a single-cell suspension of intact bacteria with ribosomal RNA (rRNA) depleted.
- Culture & Harvest: Grow bacterial culture to desired mid-log phase (OD~0.3-0.6). Harvest 10^7 - 10^8 cells by gentle centrifugation.
- Cell Wash & Resuspension: Wash cells 2x in nuclease-free 1X PBS + 0.04% UltraPure BSA. Resuspend in a suitable lysozyme-based resuspension buffer (e.g., 10mM Tris-HCl, 1mM EDTA, 1mg/ml lysozyme). Critical: Optimize lysozyme concentration and incubation time (5-15 min on ice) to permeabilize the cell wall without causing complete lysis and RNA degradation.
- rRNA Depletion: Purify total RNA using a hot phenol-chloroform method or a commercial kit designed for bacteria. Immediately treat RNA with a probe-based rRNA depletion kit (e.g., Invitrogen MICROBExpress, QIAseq FastSelect – 5S/16S/23S). Follow manufacturer protocol precisely.
- Cell Re-encapsulation: After rRNA depletion, the RNA must be re-associated with intact cells for the 10X workflow. This is the most challenging step. One approach is to re-suspend the original, permeabilized cell pellet directly in the depleted RNA supernatant after careful buffer adjustment. Alternatively, use a custom crosslinking protocol to tRNAs to cellular proteins prior to initial lysis.
B. 10X Chromium Library Construction (Day 2) Objective: Generate barcoded single-cell libraries using custom 5’ Gel Beads.
- Gel Bead Preparation: Obtain Custom 10X Gel Beads synthesized with random hexamer primers in place of poly-dT.
- Chip Loading & Partitioning: Use the Chromium Controller. Prepare the master mix per the Chromium Single Cell 5’ Reagent Kit v2 User Guide, substituting your bacterial cell + rRNA-depleted RNA sample. Target cell recovery: 5,000. Load the chip and run the controller to generate Gel Bead-in-Emulsions (GEMs).
- In-GEM Reverse Transcription (RT): Inside each GEM, random hexamers prime bacterial mRNA. The reverse transcriptase adds a template switch oligo (TSO) site, enabling full-length cDNA amplification.
- Post-GEM Cleanup & Amplification: Break emulsions, purify cDNA with DynaBeads, and amplify by PCR (12-14 cycles).
- Library Construction: Fragment, size select, and add sample indices via end-repair, A-tailing, adapter ligation, and PCR as per the 10X 5’ protocol.
C. Sequencing & Analysis (Day 3+) Objective: Sequence and demultiplex data.
- QC: Assess library size (~500-6000 bp) and concentration (qPCR).
- Sequencing: Recommended: Illumina NovaSeq or HiSeq. Read Configuration: Read1: 26 cycles (10X Barcode + UMI), i7 Index: 10 cycles, i5 Index: 10 cycles, Read2: ≥90 cycles (cDNA insert).
- Data Processing: Use Cell Ranger with a custom bacterial reference genome. Set
--chemistry SC5P-PE. Expect lower reads/cell compared to eukaryotic samples.
Visualization: The Adapted 5’ Bacterial Workflow
Title: Adapted 10X 5’ Workflow for Bacterial scRNA-seq
The Scientist's Toolkit: Key Reagents & Materials
Table 2: Essential Research Reagent Solutions
| Item | Function in Bacterial 10X Workflow |
|---|---|
| Lysozyme (Molecular Grade) | Enzymatically degrades peptidoglycan layer for gentle cell wall permeabilization, enabling RNA access without complete lysis. |
| MICROBExpress or FastSelect 5S/16S/23S Kit | Probe-based kits for selective removal of abundant ribosomal RNA (rRNA), essential for enriching bacterial mRNA prior to capture. |
| Custom 10X Gel Beads (5’) | Gel Beads containing random hexamer primers instead of poly-dT, required for priming bacterial mRNA during in-GEM reverse transcription. |
| Chromium Single Cell 5’ Reagent Kit v2 | Provides all core reagents (enzymes, buffers, primers) for library construction after GEM partitioning. The 5’ chemistry is the starting point for adaptation. |
| DynaBeads MyOne SILANE | Magnetic beads used for post-GEM cDNA cleanup and size selection, critical for removing reaction components and primer dimers. |
| TE-TW Buffer (1X TE, 0.01% Tween-20) | A gentle, nuclease-free resuspension and wash buffer for maintaining cell integrity and preventing clumping before loading. |
Application Notes
Following GEM generation and barcoding with the 10X Genomics Chromium system for bacterial single-cell RNA-seq, the workflow transitions to library preparation, quality control, and sequencing. This phase is critical for converting the barcoded cDNA into sequencer-ready libraries and ensuring data quality. A primary challenge in bacterial applications is the high ribosomal RNA (rRNA) content, which necessitates specific probe-based depletion steps not typically required in eukaryotic workflows. Key considerations include optimizing input cDNA mass, performing rigorous QC to assess library complexity and contamination, and determining sequencing depth sufficient to capture the transcriptional landscape of individual prokaryotic cells, which have lower mRNA content compared to mammalian cells.
Table 1: Key Sequencing Recommendations for Bacterial scRNA-seq on 10X Chromium
| Parameter | Typical Recommendation for Bacterial Studies | Rationale & Notes |
|---|---|---|
| Sequencing Depth | 50,000 - 100,000 reads per cell | Higher than eukaryotic standards (20-50k) to compensate for lower bacterial mRNA copy numbers and to improve detection of low-abundance transcripts. |
| Read Configuration | Paired-end (28bp Read1, 10bp i7 Index, 91bp Read2) | Read1: 16bp Chromium Barcode + 12bp UMI. Read2: Transcript sequence. 91bp Read2 is optimal for bacterial gene mapping. |
| Coverage Goal | 5-10% of the median 4 Mb bacterial genome | Aiming for sufficient transcriptome coverage per cell, though saturation is often not achieved due to technical dropout. |
| Target Cell Recovery | 5,000 - 10,000 cells | Balances library complexity with multiplet rate. For low-diversity samples, target the lower end. |
| rRNA Depletion | Essential. Use probe-based kits (e.g., Invitrogen MICROBExpress, Bioo Scientific NEXTflex RiboReduce) | Probes must be designed for the specific bacterial species or community. Performed post-cDNA amplification, pre-fragmentation. |
Experimental Protocols
Protocol 1: Post-cDNA Amplification rRNA Depletion for Bacterial scRNA-seq Libraries
This protocol is inserted between the cDNA Amplification and Library Fragmentation steps of the standard 10X Genomics Chromium Single Cell 3’ Reagent Kits v3.1.
- Material: Purified full-length cDNA from the 10X protocol.
- rRNA Depletion Kit: Use a kit compatible with your bacterial species (e.g., Thermo Fisher MICROBExpress for Bacteria).
- Procedure:
- Quantify the amplified cDNA using a fluorescence-based assay (e.g., Qubit dsDNA HS Assay).
- Use 100-500 ng of cDNA as input for the depletion reaction, following the manufacturer's instructions for hybridization conditions and incubation times.
- Perform the recommended magnetic bead-based separation to remove cDNA:rRNA probe hybrids.
- Recover the supernatant containing rRNA-depleted cDNA.
- Purify the depleted cDNA using SPRIselect beads (0.8x ratio) and elute in Tris buffer.
- QC: Analyze 1 µL on a Bioanalyzer High Sensitivity DNA chip. A successful depletion shows a reduction or elimination of the dominant rRNA-derived peak (~1.5 kb) and a more diverse smear of mRNA-derived fragments.
Protocol 2: Library Construction and Final QC
Follow the standard 10X Genomics "Library Construction" guide (Manual Part Number CG000204) with the following adjustments:
- Fragmentation, End-Repair & A-tailing: Use the entire volume of rRNA-depleted cDNA from Protocol 1 as input. The enzymatic fragmentation time (5 minutes) is fixed; do not alter.
- Sample Indexing (Dual Indexing): Use SI primers for sample multiplexing. Record index combinations meticulously.
- PCR Amplification: Perform the recommended 12-14 cycles. Quantify the final library by Qubit.
- Final Library QC:
- Fragment Size Distribution: Run 1 µL on a Bioanalyzer/ TapeStation. Expect a broad peak centered around ~450-550 bp.
- Library Molarity: Calculate using the formula: (Concentration in ng/µL * 10^6) / (library average size in bp * 650) = nM.
- qPCR for Functional Titration: Perform using the Kapa Library Quantification Kit on an Illumina platform. This is the most accurate method for determining cluster-generating molarity on flow cells.
Visualization
Diagram 1: Bacterial scRNA-seq library prep workflow
Diagram 2: How read depth impacts data quality
The Scientist's Toolkit
Table 2: Key Research Reagent Solutions for Library Prep & QC
| Item | Function in Bacterial scRNA-seq | Example Product |
|---|---|---|
| rRNA Depletion Kit | Removes abundant bacterial ribosomal RNA sequences post-cDNA amplification to enrich for mRNA. Critical for signal-to-noise ratio. | Thermo Fisher MICROBExpress, Illumina Ribo-Zero Plus |
| SPRIselect Beads | Size-selective purification of nucleic acids. Used for cleanup after cDNA amplification, rRNA depletion, and post-library construction. | Beckman Coulter SPRIselect |
| Library Quantitation Kit | Accurate quantification of final library molarity via qPCR. Essential for balanced pooling and optimal sequencer loading. | Kapa Biosystems Library Quantification Kit |
| High Sensitivity DNA Assay | Fluorometric quantification of low-concentration dsDNA samples (cDNA, final libraries). | Thermo Fisher Qubit dsDNA HS Assay |
| High Sensitivity DNA Analysis Kit | Capillary electrophoresis for assessing size distribution and quality of cDNA and final libraries. | Agilent Bioanalyzer High Sensitivity DNA Kit |
| Dual Index Kit Set A | Provides unique combinatorial indices for multiplexing up to 96 samples on Illumina sequencers. | 10X Genomics Dual Index Kit TT Set A |
This protocol details the critical data processing pipeline for single-cell RNA sequencing (scRNA-seq) of bacterial samples using the 10X Genomics Chromium platform. Within the broader thesis on adapting this technology for prokaryotic systems—which lack polyadenylated tails and have operonic gene structures—this step translates raw sequencing data into analyzable gene expression matrices. The core challenge involves modifying the standard Cell Ranger pipeline to accept custom bacterial genome references and integrating downstream tools in R/Python for microbial-specific analyses.
Key Software and Version Requirements
| Software/Tool | Version | Primary Function | Source/Link |
|---|---|---|---|
| Cell Ranger | 7.2+ | Primary alignment, barcode counting, UMI quantification | 10X Genomics Official Site |
| mkref (Cell Ranger) | Integrated | Custom reference genome construction | Bundled with Cell Ranger |
| STARsolo | 2.7.11a | Splicing-aware aligner (modified for prokaryotes) | Integrated in Cell Ranger |
| Seurat (R) | 5.1.0 | Downstream clustering, visualization, analysis | CRAN/Bioconductor |
| Scanpy (Python) | 1.10.0 | Downstream analysis in Python ecosystem | PyPI |
| Bioconductor (tximport, DESeq2) | 3.19 | Transcript-level analysis, differential expression | Bioconductor |
| Custom Python Scripts (e.g., Pandas, NumPy) | Varies | Data manipulation, custom metric calculation | PyPI |
Experimental Protocol: Constructing a Custom Reference for Prokaryotes
Objective: To build a Cell Ranger-compatible reference from a bacterial genome annotation, circumventing the need for a GTF file with standard eukaryotic features.
Materials:
- Bacterial genome FASTA file (
.fnaor.fa) - Genome annotation file (
.gffor.gff3) - High-performance computing (HPC) cluster or server with ≥32 GB RAM and 16 cores.
- Cell Ranger
mkrefpackage installed.
Procedure:
- Annotation File Conversion: Convert the bacterial GFF3 file to a modified GTF format. Prokaryotic genes must be represented as "exons" with a
gene_idandtranscript_id. Operons should be split into individual transcript entries.
- Filtering: Ensure the GTF contains only relevant features (
gene,exon). Remove tRNA, rRNA regions if analyzing mRNA only. Reference Generation: Use
cellranger mkrefwith the modified GTF.Validation: Check output
genes.gtfin the new reference directory. Verify gene counts match expectations.
Experimental Protocol: Running Cell Rangercountwith Custom Reference
Objective: To process 10X Chromium FASTQ files and generate a feature-barcode matrix for a bacterial sample.
Procedure:
- Prepare Input Files: Organize FASTQ files in the standard Cell Ranger input structure:
[Sample_Name]/[Sample_Name]_S1_L00[Lane]_[Read Type]_001.fastq.gz - Execute
cellranger count:
- Output: Key outputs include:
filtered_feature_barcode_matrix.h5: Gene-cell UMI count matrix for downstream analysis.web_summary.html: Quality control metrics (e.g., reads/cell, median genes/cell, fraction reads in cells).
Critical QC Metrics Table for Bacterial scRNA-seq:
| Metric | Target Value (Bacterial) | Interpretation | Common Issue if Off-Target |
|---|---|---|---|
| Median Genes per Cell | 500-2,000 | Transcriptome complexity | Too low: Cell lysis/poor capture. Too high: Multiplets. |
| Fraction Reads in Cells | > 70% | Specificity of capture | Low: High ambient RNA or background. |
| Estimated Number of Cells | Close to loaded | Recovery efficiency | Low: Chip failure or viability issues. |
| Reads per Cell | 20,000-100,000 | Sequencing depth | Low: Under-sequencing. High: Saturation, cost-ineffective. |
| UMI Counts per Cell | Correlates with genes | Transcript capture | Low UMI/genes: Degraded RNA or inefficient RT. |
Downstream Analysis Protocol in R (Seurat)
Objective: To perform QC, normalization, clustering, and marker gene identification on bacterial single-cell data.
Downstream Analysis Protocol in Python (Scanpy)
Objective: Equivalent analysis pipeline using the Scanpy toolkit.
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function in 10X Bacterial scRNA-seq | Example/Supplier |
|---|---|---|
| Chromium Next GEM Chip K | Partitions single bacterial cells into nanoliter-scale Gel Bead-In-Emulsions (GEMs). | 10X Genomics (1000127) |
| Chromium Next GEM Single Cell 5' Kit | Contains reagents for GEM generation, barcoding, cDNA synthesis & library construction. | 10X Genomics (1000165) |
| Prokaryotic Hybridization Wash Buffer | Custom buffer for enhancing prokaryotic mRNA capture during hybridization. | In-house formulation or NEB M-MuLV buffer |
| RNase Inhibitor (HiFi) | Protects bacterial mRNA from degradation during cell lysis and RT. | Takara Bio (2313A) |
| Murine RNase Inhibitor | Specifically inhibits common RNases, critical for low-input bacterial RNA. | NEB (M0314S) |
| Custom Template Switching Oligo (TSO) | Modified TSO to improve efficiency with bacterial mRNA lacking poly-A tails. | Integrated DNA Technologies (Custom) |
| SPRIselect Beads | Size selection and clean-up of cDNA and final libraries. | Beckman Coulter (B23318) |
| Dual Index Kit TT Set A | Provides unique dual indices for sample multiplexing. | 10X Genomics (1000215) |
Workflow and Data Analysis Diagrams
Diagram Title: Full scRNA-seq Pipeline from FASTQ to Insights
Diagram Title: Building a Custom Bacterial Reference for Cell Ranger
Solving Common Pitfalls: Optimizing Bacterial scRNA-seq Experiments on the 10X Platform
Effective single-cell RNA sequencing (scRNA-seq) of bacterial populations using the 10X Genomics Chromium platform presents a unique challenge due to the fundamental structural differences between prokaryotic and eukaryotic cells. A core thesis of adapting Chromium technology for bacterial research is that the standard chemical lysis protocols optimized for mammalian cells are insufficient for robust bacterial cell wall disruption, particularly for Gram-positive species. This inefficiency leads to low RNA capture efficiency, skewing transcriptomic data and limiting the detection of low-abundance transcripts. This application note details optimized, species-specific permeabilization and lysis strategies to achieve robust and reproducible bacterial single-cell transcriptomes, enabling the study of microbial heterogeneity, antibiotic persistence, and host-pathogen interactions at unprecedented resolution.
The Challenge: Bacterial Cell Wall Architecture
The primary barrier to efficient RNA capture is the bacterial cell wall. Gram-negative bacteria possess a thin peptidoglycan layer surrounded by an outer membrane containing lipopolysaccharide (LPS). Gram-positive bacteria have a thick, multi-layered peptidoglycan sacculus with teichoic acids. These structures are highly resistant to standard lysis buffers.
Table 1: Key Structural Differences Impacting Lysis
| Feature | Gram-negative Bacteria | Gram-positive Bacteria |
|---|---|---|
| Peptidoglycan Layer | Thin (2-7 nm) | Thick (20-80 nm) |
| Outer Membrane | Present (LPS) | Absent |
| Permeability Barrier | Outer Membrane | Peptidoglycan & Cell Membrane |
| Primary Lysis Target | Outer Membrane & Peptidoglycan | Peptidoglycan & Cell Membrane |
| Relative Lysis Difficulty | Moderate | High |
Optimized Permeabilization & Lysis Protocols
The following protocols are designed to be integrated upstream of the 10X Genomics Chromium Next GEM chip loading and library preparation workflow. Cell viability and integrity must be confirmed prior to processing.
General Preparation & Common Reagents
- Wash Buffer: 1X PBS, 0.04% UltraPure BSA, 1U/µl RNase Inhibitor.
- Resuspension Buffer: Nuclease-free water or TE buffer with 0.04% BSA and 1U/µl RNase Inhibitor.
- Goal: Achieve a single-cell suspension at the target cell concentration (e.g., 500-1,200 cells/µl for Chromium Next GEM).
Protocol A: Gram-negative Species (e.g.,E. coli,P. aeruginosa)
This protocol uses Lysozyme to degrade peptidoglycan, followed by a mild detergent to solubilize membranes.
Workflow Diagram:
Diagram Title: Gram-negative Bacterial Lysis for 10X
Detailed Steps:
- Lysis Buffer Preparation: 10 mM Tris-HCl (pH 8.0), 0.1% N-Lauroylsarcosine, 5 mM EDTA, 1U/µl RNase Inhibitor. Add Lysozyme (1 mg/ml final concentration) fresh.
- Pellet 1x10⁶ - 1x10⁷ bacterial cells, wash twice in ice-cold Wash Buffer.
- Gently resuspend cell pellet in 100 µl of prepared Lysis Buffer. Do not vortex.
- Incubate on ice for 5 minutes. Monitor under microscope for loss of cell refractility.
- Immediately dilute the reaction with 900 µl of ice-cold Wash Buffer containing 2U/µl RNase Inhibitor to quench lysis.
- Keep on ice for 2 minutes.
- Pass through a 30 µm flow filter to remove debris and aggregates.
- Perform cell count and viability check (if using a viability dye compatible with downstream steps). Adjust concentration for Chromium.
Protocol B: Gram-positive Species (e.g.,S. aureus,B. subtilis)
This sequential protocol uses enzymatic weakening of the peptidoglycan layer followed by mechanical disruption.
Workflow Diagram:
Diagram Title: Gram-positive Bacterial Lysis for 10X
Detailed Steps:
- Enzymatic Pre-treatment: Wash cell pellet (1x10⁷ cells) and resuspend in 100 µl of PBS with species-specific enzyme.
- For S. aureus: Use Lysostaphin (50 µg/ml final).
- For B. subtilis: Use Lysozyme (2 mg/ml final) with 10 mM EDTA.
- Add 1U/µl RNase Inhibitor.
- Incubate at 37°C for 10 minutes. This creates "protoplasts" or weakened cells.
- Centrifuge gently (2000 x g, 4°C, 2 min). Remove supernatant.
- Resuspend pellet in 100 µl of Mild Lysis Buffer (0.1% Triton X-100, 10 mM Tris, 1U/µl RNase Inhibitor).
- Apply Controlled Mechanical Disruption:
- Option 1 (Sonication): Use a microtip sonicator. 3 pulses of 5 seconds at 10% amplitude, on ice. Allow 30-second cooling between pulses.
- Option 2 (French Press): Pass cells once at 6,000 psi.
- Immediately transfer to tube with 900 µl ice-cold Wash Buffer + Inhibitor.
- Quench on ice for 5 min.
- Filter through a 30 µm filter. Count and adjust concentration. Avoid over-sonication.
Table 2: Performance Metrics of Optimized Protocols vs. Standard 10X Lysis
| Metric | Standard 10X Lysis (Eukaryotic) | Protocol A (Gram-negative) | Protocol B (Gram-positive) |
|---|---|---|---|
| Estimated Lysis Efficiency | <20% (Gram-neg), <5% (Gram-pos) | 85-95% | 70-90% (varies by species) |
| RNA Molecule Recovery per Cell | Very Low / Undetectable | High (5,000-15,000) | Moderate-High (3,000-10,000) |
| Gene Detection per Cell | <100 | 1,000-2,500 | 800-2,000 |
| Cell Throughput (Recovered) | Low due to intact cell filter | High | Moderate-High |
| Key Risk | Data from lysed sub-population only | Over-lysis & RNA degradation | Complete RNA fragmentation (if over-processed) |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Bacterial scRNA-seq Lysis
| Reagent / Kit | Function in Protocol | Key Consideration |
|---|---|---|
| Lysozyme (from chicken egg white) | Degrades peptidoglycan layer by hydrolyzing β-(1,4) linkages. | Use molecular biology grade. Concentration is critical (1-2 mg/ml). |
| Lysostaphin | Specifically cleaves pentaglycine bridges in S. aureus peptidoglycan. | Essential for robust S. aureus lysis. Ineffective against other species. |
| N-Lauroylsarcosine (Sarkosyl) | Ionic detergent for solubilizing Gram-negative outer and inner membranes. | Harsher than Triton X-100; use at low concentration (0.1%). |
| Triton X-100 | Non-ionic detergent for gentle membrane permeabilization. | Used for Gram-positives after enzymatic weakening. |
| RNase Inhibitor (e.g., Murine) | Inactivates RNases released during cell disruption. | Must be added to ALL buffers post-wash. Critical for RNA integrity. |
| EDTA (Ethylenediaminetetraacetic acid) | Chelates Mg²⁺, destabilizing the outer membrane and inhibiting DNases/RNases. | Enhances lysozyme activity, especially for Gram-positives. |
| 10X Genomics Chromium Next GEM Kit | Provides microfluidic partitioning, barcoding, and library prep reagents. | The optimized lysis protocol feeds directly into the "Cell Suspension" step of this kit. |
| 30 µm Cell Strainer (Flow filter) | Removes cellular debris and aggregates prior to loading chip. | Prevents clogging of microfluidic channels on the Chromium chip. |
| UltraPure BSA | Stabilizes cells and blocks non-specific binding. | Used in wash buffers to maintain cell suspension and viability. |
Critical Considerations & Troubleshooting
- Titration is Essential: The optimal enzyme concentration and incubation time are species and strain-dependent. Perform a lysis efficiency test (microscopy, viability stain) before a full Chromium run.
- Temperature Control: All steps after enzyme/detergent addition must be performed on ice or at 4°C unless incubating, to slow RNase activity.
- Quenching: Immediate dilution into a large volume of cold, inhibitor-rich buffer is as important as the lysis step itself.
- Quality Control: Post-lysis, check for the presence of intact cells (which will partition but yield no data) and assess RNA integrity from a bulk sample if possible.
- Integration with 10X Workflow: The final cell suspension in Wash Buffer must be compatible with the 10X Chemistry. Avoid introducing contaminants or enzymes (e.g., active Proteinase K) into the chip.
By implementing these species-optimized permeabilization and lysis protocols, researchers can overcome the central bottleneck of low RNA capture efficiency, thereby unlocking the potential of the 10X Genomics Chromium platform for robust and high-resolution single-cell transcriptomic analysis of both Gram-positive and Gram-negative bacteria.
Within a broader thesis employing the 10X Genomics Chromium platform for bacterial single-cell RNA sequencing (scRNA-seq), a paramount challenge is the management of high background and ambient RNA. Bacterial transcripts are predominantly ribosomal RNA (rRNA), often constituting >80% of total RNA. In droplet-based workflows, lysed cell debris and free RNA molecules contribute to ambient background, obscuring true single-cell transcriptomes. Effective rRNA depletion and controlled nuclease treatment are therefore critical pre-processing steps to enhance mRNA capture, improve library complexity, and yield biologically meaningful data on bacterial heterogeneity, antibiotic responses, and virulence pathways in host-relevant contexts.
Quantitative Comparison of Depletion Strategies
Table 1: Comparison of Bacterial rRNA Depletion Methods
| Method | Principle | Approximate rRNA Reduction | Compatible with 10X 3’ Gene Expression | Input RNA Requirement | Key Considerations |
|---|---|---|---|---|---|
| Commercial Probe-Based (Ribo-Zero) | DNA probe hybridization to rRNA followed by removal. | 85-95% | Yes, post-lysis. | 1-1000 ng total RNA | Can deplete multiple rRNA types; may require DNase step. |
| RNase H-based Depletion | rRNA-specific oligonucleotides guide RNase H to cleave rRNA. | >90% | Yes, post-lysis. | 10-1000 ng total RNA | Highly specific; effective for diverse bacterial species. |
| 5’ Nuclease Treatment (Duplex-Specific Nuclease - DSN) | Degrades ds cDNA/duplexed rRNA sequences post-reverse transcription. | Up to 80% | Limited, can affect mRNA if not carefully optimized. | Varies | Requires careful temperature/enzyme concentration control. |
| mRNA Enrichment by Poly-A Selection | Oligo-dT capture of polyadenylated mRNA. | Ineffective for most bacterial mRNA | No | N/A | Most bacterial mRNAs lack poly-A tails; not recommended. |
| Custom CRISPR/Cas-based Depletion | Cas13a RNA-guided degradation of specific rRNA sequences. | >95% (in development) | Potentially, post-lysis. | Research phase | High specificity and programmability for novel species. |
Table 2: Impact of Ambient RNA Reduction Treatments on 10X Data Metrics
| Treatment | Protocol Point | Effect on Median Genes/Cell (Bacteria) | Effect on Ambient RNA Contamination (Soup%) | Effect on rRNA Reads (%) | Recommended for Low-Biomass Samples? |
|---|---|---|---|---|---|
| No Treatment (Control) | N/A | Baseline (e.g., 500 genes) | High (e.g., 25%) | >70% | No |
| Pre-lysis Probe Depletion (Ribo-Zero) | Before GEM generation | Increase of 30-50% | Moderate reduction (to ~15%) | <15% | Yes, but may lose some cell integrity. |
| Post-lysis Nuclease Treatment (within GEMs) | During RT or after lysis in droplets | Increase of 20-40% | Significant reduction (to <10%) | <20% | Yes, compatible with standard workflow. |
| External Nuclease (e.g., Benzonase) Wash | Prior to loading on Chromium | Minimal increase | Potentially high reduction of ambient RNA | Minimal direct impact | Yes, for reducing extracellular background. |
| Bioinformatic Soup Correction (CellBender) | Post-sequencing | Maintains count | Corrects contamination computationally | No direct impact | Yes, as a complementary step. |
Detailed Experimental Protocols
Protocol 3.1: Integrated RNase H-based rRNA Depletion for 10X Bacterial scRNA-seq
Objective: To deplete rRNA from bacterial lysates prior to cDNA amplification within the 10X Chromium workflow.
Materials: See "The Scientist's Toolkit" below. Procedure:
- Culture and Harvest: Grow bacterial culture to mid-log phase. Harvest 1x10^6 - 1x10^8 cells by gentle centrifugation.
- Gentle Lysis: Resuspend cell pellet in 100 µl of 10X Lysis Buffer (supplemented with 1% β-mercaptoethanol). Incubate at 25°C for 5 minutes. Immediately place on ice.
- rRNA Hybridization: To the lysate, add 5 µl of a pooled, species-specific anti-rRNA DNA oligonucleotide set (100 µM total). Heat at 70°C for 5 min, then gradually cool to 45°C over 20 min.
- RNase H Digestion: Add 2 µl of RNase H (5 U/µl) and 12 µl of 10X RNase H Buffer. Incubate at 45°C for 30 min.
- Purification: Purify the RNA using RNAClean XP beads at a 1.8x ratio. Elute in 15 µl of Nuclease-Free Water.
- Quality Assessment: Run 1 µl on a Bioanalyzer RNA Pico Chip to confirm rRNA peak reduction.
- 10X Library Construction: Proceed immediately with the purified RNA using the 10X Chromium Single Cell 3’ Reagent Kit (v3.1 or later), starting at the RT Master Mix step. Adjust input volume to a maximum of 14.5 µl.
Protocol 3.2: Post-Lysis Nuclease Treatment in Droplets for Ambient RNA Reduction
Objective: To degrade ambient RNA co-encapsulated in droplets without damaging intracellular mRNA.
Materials: See "The Scientist's Toolkit" below. Procedure:
- Master Mix Preparation: Prepare the Reverse Transcription Master Mix as per 10X protocol, but supplement it with 0.05 U/µl of Exonuclease I (Exo I) and 0.01 U/µl of RNase A.
- Rationale: Exo I degrades single-stranded DNA primers not incorporated, reducing background. RNase A, at very low concentration, preferentially degrades exposed RNA fragments (ambient RNA) before the cell lysis and RT reagents fully activate.
- GEM Generation and Incubation: Generate Gel Bead-in-Emulsions (GEMs) as per manufacturer's instructions. Immediately after GEM generation, perform a brief incubation at 25°C for 2 minutes before transferring to the PCR thermal cycler.
- Thermal Cycling: Place GEMs in a pre-warmed PCR block at 53°C and run the standard 10X RT program (45 min at 53°C). The initial low-temperature step allows nuclease activity before the protective environment of reverse transcription is fully established for intact mRNA.
- Post-RT Cleanup: Proceed with the standard Silane bead cleanup to remove enzymes and reaction components.
- Optimization Note: The concentration of RNase A is critical. A titration (0.005 - 0.05 U/µl) is recommended for each new bacterial application.
Visualizations
Title: Integrated Wet-Lab scRNA-seq Workflow for Bacteria
Title: Problem-Strategy-Outcome Logic for rRNA & Ambient RNA
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions
| Item | Function in Protocol | Example Product/Kit |
|---|---|---|
| Species-specific Anti-rRNA Oligo Pool | Hybridizes to 16S, 23S, 5S rRNA for targeted depletion. | Custom-designed DNA oligos (IDT); Ribo-Zero rRNA Removal Kit (Bacteria) |
| RNase H | Enzyme that cleaves RNA in RNA-DNA hybrids, critical for probe-based depletion. | E. coli RNase H (NEB) |
| Exonuclease I (Exo I) | Degrades residual single-stranded DNA primers to reduce sequencing background. | Exonuclease I (NEB) |
| Controlled RNase A | At low concentration, degrades exposed ambient RNA fragments in droplets. | RNase A, Molecular Grade (Thermo) |
| RNAClean XP Beads | SPRI bead-based purification for RNA cleanup and size selection post-depletion. | AMPure XP/RNAClean XP (Beckman Coulter) |
| 10X Lysis Buffer | Part of 10X kit; gently lyses bacterial cells while preserving RNA integrity. | Single Cell 3’ Reagent Kits (10X Genomics) |
| Bioanalyzer/Pico Chip | Microfluidics-based system to assess RNA quality and confirm rRNA depletion. | Agilent 2100 Bioanalyzer with RNA Pico Kit |
| Cell-Friendly Lysis Additive | Enhances bacterial cell wall lysis without degrading RNA (e.g., lysozyme, mutanolysin). | Lysozyme (Sigma); Mutanolysin (Sigma) |
| Duplex-Specific Nuclease (DSN) | Alternative enzyme for normalizing cDNA by degrading abundant dsDNA/RNA duplexes. | DSN Enzyme (Evrogen) |
| Bioinformatic Tool: CellBender | Computational removal of ambient RNA contamination from count matrices. | CellBender (GitHub) |
Cell Multiplexing and Doublet Detection in Bacterial Populations Using Cell Hashing or Genetic Barcodes
Single-cell RNA sequencing (scRNA-seq) of bacterial populations presents unique challenges, including limited starting RNA, the need to differentiate individual cells in communities, and the pervasive risk of doublets or multiplets that confound data analysis. Within the framework of the 10X Genomics Chromium platform—primarily designed for eukaryotic cells—adapting these workflows for prokaryotes requires specialized sample multiplexing and doublet detection strategies. Cell hashing and genetic barcoding are two pivotal techniques that enable sample pooling, increase throughput, reduce costs, and provide a robust mechanism for doublet identification in bacterial single-cell studies. This application note details the protocols and considerations for implementing these methods in bacterial research using the 10X Chromium system.
Core Principles and Adaptation for Bacteria
The 10X Chromium system relies on Gel Bead-In-EMulsions (GEMs) to partition single cells. Each gel bead is coated with oligonucleotides containing a unique barcode for cell identification, a Unique Molecular Identifier (UMI), and a poly-dT sequence for mRNA capture. Since bacterial mRNA lacks polyadenylated tails, a polyadenylation step or the use of random primers is required—a critical pre-processing modification.
Cell hashing uses antibody-conjugated oligonucleotide tags (Hashtags) that bind to ubiquitous surface proteins, labeling cells from different samples with distinct barcodes before pooling. Genetic barcoding involves the stable integration of unique DNA sequences into the genome of distinct bacterial strains or populations prior to the experiment. In both cases, after sequencing, the hashtag or genetic barcode reads are used to demultiplex samples and identify droplets containing cells from more than one sample (inter-sample doublets).
Key Research Reagent Solutions
| Reagent / Material | Function in Bacterial scRNA-seq | Key Consideration for Prokaryotes |
|---|---|---|
| 10X Chromium Controller & Chip | Generates single-cell GEMs. | Standard hardware; protocol modifications occur upstream. |
| Chromium Next GEM Single Cell 3’ Reagent Kit | Provides reagents for GEM generation, RT, cDNA amplification & library construction. | Requires upstream bacterial cell wall treatment and mRNA enrichment/polyadenylation. |
| Cell Hashing Antibodies (e.g., TotalSeq-A/B/C) | Conjugated to oligonucleotide hashtags for sample multiplexing. | Must bind to conserved bacterial surface epitopes (e.g., outer membrane proteins). May require species-specific validation. |
| Genetic Barcoding Plasmid Library | Introduces heritable, diverse oligonucleotide sequences into bacterial genomes. | Requires efficient transformation/conjugation system for target species. Barcode must be expressed and captured during library prep. |
| Poly(A) Tailing Kit | Adds poly-A tails to bacterial 3’ ends of RNAs. | Critical for capture by Chromium poly-dT beads. Must optimize reaction time to avoid excessive tailing. |
| Bacterial Lysis & RNA Stabilization Buffer | Gently breaks cell wall without degrading RNA. | Must be compatible with 10X RT master mix. Lysozyme concentration is critical. |
| RNase Inhibitor | Protects bacterial mRNA during processing. | Essential due to high RNase activity in bacterial lysates. |
| Magnetic mRNA Isolation Beads | Enriches for mRNA prior to poly-A tailing. | Removes ribosomal RNA which constitutes >95% of bacterial total RNA. |
Detailed Experimental Protocols
Protocol 4.1: Genetic Barcoding and Sample Preparation
Objective: Generate genetically distinct, heritably barcoded bacterial populations for pooling.
- Barcode Library Construction: Clone a diverse library of 12-16bp random oligonucleotides, flanked by constant primer binding sites, into a neutral genomic site or a plasmid with moderate copy number in your target bacterial species. Ensure the barcode will be transcribed.
- Strain Generation: Transform or conjugate the barcode library into your bacterial strain of interest. Plate to obtain individual colonies.
- Population Expansion: Pick multiple colonies (each representing a unique barcode) and grow them in separate cultures under identical conditions to the desired OD600.
- Sample Processing: For each culture, harvest cells. Gently wash 2x with nuclease-free PBS. Proceed to cell hashing (if combining with hashtags) or direct fixation/permeabilization for single-cell processing.
Protocol 4.2: Cell Hashing of Bacterial Samples
Objective: Label distinct bacterial samples with oligonucleotide-conjugated antibodies for multiplexing.
- Antibody Titration: Titrate the candidate hashtag antibody on your bacterial cells to determine the optimal concentration that maximizes signal without clumping. Note: Many commercial hashtag antibodies target human CD proteins. For bacteria, antibodies against common surface factors (e.g., E. coli OmpA) must be validated and conjugated to hashtag oligonucleotides in-house or sourced custom.
- Cell Staining: Resuspend each bacterial pellet (1-5x10^5 cells) in 50-100µL of PBS containing the optimized concentration of hashtag antibody.
- Incubation: Incubate on ice or at 4°C for 30 minutes with gentle agitation.
- Washing: Wash cells 3x with excess PBS + 0.04% BSA to remove unbound antibody.
- Pooling: Count cells from each hashtag-labeled sample and pool them at equal numbers into a single tube. Keep on ice.
Protocol 4.3: 10X Chromium Library Preparation for Multiplexed Bacterial Samples
Objective: Generate single-cell RNA-seq libraries from pooled, barcoded bacterial samples.
- Cell Preparation: For genetically barcoded or hashtagged pools, determine total cell count and viability. Adjust concentration to 700-1200 cells/µL in nuclease-free PBS or resuspension buffer.
- Critical Lysis Optimization: For Gram-negative bacteria, add lysozyme (0.1-1.0 mg/mL final) to the master mix. For Gram-positive, consider a mild permeabilization step (e.g., 0.01% Triton X-100) prior to loading. Validate lysis efficiency.
- mRNA Capture & Reverse Transcription: Load the cell suspension onto the 10X Chromium chip per manufacturer's instructions. Within the GEMs, bacterial cells are lysed, releasing RNA.
- Poly-A Capture Modification: Since bacterial mRNA lacks poly-A tails, one of two modifications is required:
- A. Pre-GEM Polyadenylation: Prior to loading cells, isolate total RNA, enrich for mRNA, perform in vitro poly(A) tailing, and then load the RNA (not cells) onto the Chromium system. This loses cellular resolution but captures transcriptomes.
- B. Post-Lysis Polyadenylation in GEMs: Use gel beads coated with poly-dT and a terminal transferase enzyme within the GEM RT mix to polyadenylate bacterial RNA immediately prior to capture and RT. This is an experimental workflow requiring custom bead synthesis.
- Poly-A Capture Modification: Since bacterial mRNA lacks poly-A tails, one of two modifications is required:
- Post-GEM Processing: Continue with standard cDNA amplification and library construction steps as per the Chromium Single Cell 3' Reagent Kit v3.1 User Guide.
- Hashtag/Genetic Barcode Library Construction: A separate PCR reaction is performed using a small aliquot of amplified cDNA (or a separate sample index PCR for feature barcoding) to enrich for the hashtag or genetic barcode sequences, creating a separate sequencing library.
- Sequencing: Pool the gene expression library and the barcode library. Sequence on an Illumina system. The gene expression library typically requires ~50,000 reads/cell, while the barcode library requires far fewer (~5,000 reads/cell).
Data Analysis & Doublet Detection Workflow
The primary analysis leverages the Cell Ranger (10X Genomics) pipeline with additional tools for multiplexing analysis.
- Alignment & Counting: Use
cellranger countwith a custom reference genome containing the genetic barcode sequence (if applicable) to generate a feature-barcode matrix. - Sample Demultiplexing:
- For Cell Hashing: Use
CITE-seq-CountorCell Ranger's feature barcoding pipeline to count hashtag oligonucleotide (HTO) reads per cell barcode. - For Genetic Barcoding: Extract reads aligning to the constant barcode region and call the variable barcode sequence.
- For Cell Hashing: Use
- Doublet Identification:
- Cells are classified based on their HTO or genetic barcode signal. Cells with high counts for more than one barcode are classified as inter-sample doublets.
- Tools like DemuxEM, HTODemux, or Seurat's
HTODemux()function are used for hashtag data. Genetic barcodes are demultiplexed using tools like GMM-Demux or Vireo. - Intra-sample doublets (multiple cells of the same sample in one droplet) must be identified bioinformatically using tools like DoubletFinder or scrublet, which look for anomalous gene expression profiles.
Table 1: Quantitative Metrics for Doublet Detection Methods in Bacterial Pools
| Method | Principle | Detection Rate* | Key Advantage for Bacteria | Required Sequencing Depth (Barcode) |
|---|---|---|---|---|
| Cell Hashing (HTO) | Surface-protein binding oligonucleotide tags. | 1-5% of loaded cells | Can be used on wild-type, non-engineered strains. | Low (~1-5k reads/cell) |
| Genetic Barcoding | Heritable genomic DNA barcode. | <1% (if clonal populations are pure) | Permanent, stable label; no staining step. | Low (~1-5k reads/cell) |
| Bioinformatic (DoubletFinder) | Artificial nearest-neighbor profile prediction. | Varies with cell number | Identifies intra-sample doublets missed by multiplexing. | N/A (uses gene expression) |
*Doublet rate is highly dependent on cell loading concentration on the Chromium.
Workflow and Pathway Visualizations
Title: Bacterial scRNA-seq Multiplexing and Doublet Detection Workflow
Title: GEM Scenarios: Singlets vs. Inter-Sample Doublets
Within the broader thesis on optimizing 10X Genomics Chromium workflows for bacterial single-cell RNA sequencing (scRNA-seq), maintaining high cell viability and ensuring accurate recovery post-processing are paramount. Bacterial cells present unique challenges due to their size, cell wall structure, and sensitivity to osmotic stress. This application note details the critical pre-Chromium steps of washing and resuspension that directly impact final library quality, data fidelity, and the success of downstream drug discovery applications.
Key Challenges and Quantitative Impact
Improper handling during washing and resuspension leads to cell aggregation, lysis, and transcriptional stress responses, which introduce significant noise in scRNA-seq data. The following table summarizes the quantitative impact of key parameters on cell viability and recovery, as established in recent literature and optimized protocols.
Table 1: Impact of Processing Parameters on Bacterial Cell Viability and Recovery
| Parameter | Suboptimal Condition | Optimal Condition | Typical Viability Impact (vs. Optimal) | Data Quality Risk |
|---|---|---|---|---|
| Wash Buffer Osmolarity | DI Water or Standard PBS (~300 mOsm) | Iso-osmotic Buffer (e.g., ~800-1000 mOsm for many bacteria) | Decrease of 40-60% | High cell lysis, background RNA contamination. |
| Centrifugation Force | >5000 x g | 300-500 x g (for pelleted cultures) | Decrease of 20-40% | Cell clumping, physical damage, loss of sensitive phenotypes. |
| Resuspension Buffer | Growth Media or Simple Saline | Specified Dilution Buffer (e.g., 10X Diluent C) | Decrease of 30-50% | Poor droplet formation, low cell recovery in Chromium. |
| Temperature during Wash | Room Temperature or 4°C | Consistently 4°C | Decrease of 10-20% | Induction of cold-shock stress response genes. |
| Resuspension Pipetting | Vortexing or vigorous pipetting | Gentle wide-bore pipette tip aspiration | Decrease of 15-30% | Shear stress, cell wall damage, aggregation. |
Detailed Protocols
Protocol 3.1: Gentle Wash for Pelleted Bacterial Cultures
Objective: To remove spent growth media and inhibitors while maximizing cell viability. Materials: See "The Scientist's Toolkit" below. Procedure:
- Harvesting: Pellet 1-5 x 10^8 cells from mid-log phase culture by centrifugation at 300-500 x g for 5-10 minutes at 4°C.
- Supernatant Removal: Carefully decant or aspirate supernatant, leaving ~50 µL to avoid disturbing the pellet.
- First Wash: Gently resuspend the pellet in 1 mL of ice-cold, iso-osmotic wash buffer (e.g., 1X PBS supplemented with 0.9M sucrose or appropriate osmolyte) using a wide-bore 1000 µL pipette tip. Do not vortex.
- Re-pellet: Centrifuge at 300-500 x g for 5 minutes at 4°C.
- Second Wash & Final Pellet: Repeat steps 2-4.
- Final Aspiration: Aspirate supernatant completely using a fine-tip pipette.
Protocol 3.2: Optimized Resuspension for 10X Chromium Loading
Objective: To achieve a single-cell suspension at the correct concentration in a buffer compatible with the Chromium chip. Materials: See "The Scientist's Toolkit" below. Procedure:
- Pre-wet Tip: Pre-wet a wide-bore pipette tip with the target resuspension buffer (e.g., 10X Genomics "Diluent C" or validated custom buffer).
- Initial Resuspension: Add 10-20 µL of buffer to the washed cell pellet. Gently pipette up and down 5-10 times until no visible clumps remain. Avoid introducing air bubbles.
- Volume Adjustment: Bring the suspension to the desired final volume (typically 30-100 µL) with additional buffer. The target concentration is ~1,000-10,000 cells/µL, but must be optimized empirically for each bacterial strain and growth condition.
- Filtration (Critical): Pass the entire suspension through a pre-wetted, cell strainer cap (e.g., 20-40 µm mesh) into a sterile, low-binding microcentrifuge tube.
- Viability Assessment: Mix 10 µL of cell suspension with 10 µL of acridine orange/propidium iodide (AO/PI) or equivalent viability dye. Count using a fluorescence microscope or automated cell counter. Target viability >90%.
- Loading: Keep the filtered suspension on ice and load into the Chromium chip within 15 minutes.
Visualized Workflows
Workflow for Optimal Bacterial Cell Prep
Impact of Poor Handling on scRNA-seq Data
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for Bacterial scRNA-seq Prep
| Item | Function & Rationale | Example Product/Composition |
|---|---|---|
| Iso-osmotic Wash Buffer | Maintains osmolarity close to bacterial cytosol to prevent lysis and osmotic shock. | 1X PBS with 0.9M Sucrose (adjust molarity for species). |
| Chromium-compatible Dilution Buffer | Ensures cell suspension chemistry is optimal for droplet generation and barcoding in the Chromium controller. | 10X Genomics "Diluent C" (validated for system). |
| RNase Inhibitors | Protects released RNA from degradation during processing, crucial after cell wall disruption. | Recombinant RNase Inhibitor (e.g., RNasin Plus). |
| Wide-Bore/Low-Binding Pipette Tips | Minimizes shear stress during resuspension and prevents cell adhesion to tip walls, aiding accurate recovery. | Sterile, aerosol-barrier tips with >1 mm orifice. |
| Cell Strainer Caps (20-40 µm) | Removes cell aggregates and debris that would clog the Chromium chip microfluidics. | Flowmi or PluriSelect strainer caps. |
| Fluorescent Viability Stain | Accurately discriminates live/dead cells for precise concentration calculation and viability QC. | AO/PI, SYTO BC/Propidium Iodide dual stains. |
| Pre-chilled, Low-Binding Microtubes | Maintains sample at 4°C, reduces cell adhesion to tube walls, maximizing recovery. | PCR tubes or microtubes made of polypropylene. |
Within a thesis focused on leveraging the 10X Genomics Chromium platform for bacterial single-cell RNA sequencing (scRNA-seq) research, a paramount bioinformatic challenge is the accurate discrimination of genuine bacterial transcriptional signals. This process is confounded by overwhelming host-derived mRNA contamination and technical noise inherent in low-biomass applications. Effective filtering is critical for downstream analyses, including the identification of bacterial transcriptional states within host niches and the discovery of potential therapeutic targets.
Core Computational Filtering Strategies
Hierarchical Filtering Workflow
The recommended bioinformatic pipeline employs a sequential, hierarchical approach to incrementally refine the data.
Diagram Title: Hierarchical Bioinformatic Filtering Workflow
Quantitative Filtering Benchmarks
The following table summarizes typical thresholds and metrics used at key filtering stages, derived from recent methodological studies.
Table 1: Key Filtering Parameters and Benchmarks
| Filtering Stage | Tool/Technique | Key Parameter | Typical Threshold / Action | Primary Target |
|---|---|---|---|---|
| Host Read Depletion | KneadData (Kraken2), BBsplit, STAR | Host genome alignment rate | >95% of reads removed; retain <5% host alignment | Eukaryotic host mRNA |
| Bacterial Alignment | STAR (with prokaryotic settings), Bowtie2, BWA | Unique mapping rate | Aim for >70% uniquely mapped to bacterial reference(s) | Non-specific/ multi-mapped reads |
| Ambient RNA Removal | CellBender, DecontX, SoupX, EmptyDrops | Contamination fraction | Estimate and subtract; often 1-20% of counts per cell are ambient | Background free RNA |
| Expression Threshold | Seurat, Scanpy, custom scripts | Minimum genes per bacterial "cell" | >5 bacterial genes/cell (highly variable based on infection model) | Empty droplets / low-quality events |
| Noise Reduction | Scrublet, DoubletFinder | Doublet prediction score | Remove top 5-10% predicted doublets | Multiple bacteria in one partition |
Detailed Experimental Protocols
Protocol 1: In Silico Host Read Depletion and Bacterial Alignment for 10X Data
Objective: To separate sequencing reads derived from the host genome from those originating from bacterial transcripts.
Materials: High-performance computing cluster, 10X Cell Ranger FASTQ outputs, host reference genome (e.g., GRCh38), bacterial reference genome(s).
Procedure:
- Concatenate FASTQs: Combine lanes if necessary using
cat. - Host Depletion with KneadData:
- Bacterial Alignment with STAR: STAR must be used in "GeneCounts" mode with a prokaryotic GTF.
- Generate Count Matrix: The
ReadsPerGene.out.tab file from STAR is parsed to create a feature-barcode matrix compatible with downstream single-cell analysis tools (e.g., Seurat).
Protocol 2: Ambient RNA Removal Using CellBender
Objective: To statistically model and subtract background RNA contamination shared across all droplets.
Procedure:
- Prepare Input: Generate a raw H5 count matrix (feature-barcode matrix) from Cell Ranger or your bacterial alignment pipeline.
- Run CellBender remove-background:
- Output Validation: Load the
corrected_matrix.h5 and compare the number of genes per cell and total counts before and after correction. A successful run typically shows reduced background without eliminating signal from low-input cells.
Protocol 3: Doublet Detection in Bacterial scRNA-seq
Objective: To identify and remove droplets containing transcripts from more than one bacterial cell, which can create artificial transcriptional profiles.
Procedure (using Scrublet in Python):
Filter the bacterial count matrix using the singlet_mask before proceeding to clustering and differential expression.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents and Materials for Bacterial 10X scRNA-seq
Item
Function in Bacterial scRNA-seq
Key Consideration
10x Genomics Chromium Controller & Kits
Partitions individual bacterial cells (or transcripts) into nanoliter-scale Gel Beads-in-emulsion (GEMs).
Use Single Cell 3' Gene Expression v3.1 kit. Critical: No rRNA depletion step is used for bacteria.
Prokaryotic Lysis Buffer
Chemically disrupts the tough bacterial cell wall to release RNA.
Must be compatible with 10X RT mix. Optimization for Gram-positive/-negative is required (e.g., lysozyme + proteinase K).
Poly-A Carrier RNA
Improves cDNA recovery and library yield from low-input bacterial samples.
Mitigates losses during library prep; does not interfere with bacterial transcript capture.
ERCC (External RNA Controls Consortium) Spike-in
Inert synthetic RNA molecules added to the lysis buffer for technical noise assessment.
Distinguishes true biological variation from technical dropouts. Essential for low-input work.
RNase Inhibitor
Protects released bacterial mRNA from degradation during processing.
Use a heat-stable, potent inhibitor to maintain RNA integrity during lysis and RT.
Custom Gel Beads
Contain poly-dT primers for cDNA synthesis.
Standard beads target eukaryotic poly-A tails. For bacterial poly-A- transcripts, beads must be custom-designed with gene-specific or random hexamer primers.
Dual-Indexed Library Preparation Kit
Adds sample indices and P5/P7 adapters for sequencing.
Follow 10X protocol. Dual indexing is critical for multiplexing multiple infection conditions.
PhiX Control v3
Spiked into sequencing run for low-diversity library calibration.
Bacterial libraries often have lower transcriptome complexity than eukaryotic, making PhiX essential for cluster detection on Illumina flow cells.
Pathway: Decision Logic for Filter Acceptance
The final step involves a logical assessment to classify a droplet/barcode as containing a legitimate bacterial transcriptional profile.
Diagram Title: Decision Logic for Accepting a Bacterial Transcriptome
Benchmarking Performance: Validating 10X Chromium Data Against Bulk RNA-seq and Alternative scRNA Platforms
Within a thesis investigating host-pathogen interactions using 10X Genomics Chromium for bacterial single-cell RNA sequencing (scRNA-seq), robust validation is paramount. The platform's droplet-based methodology can capture transcriptional profiles of individual bacteria-infected cells or even bacteria themselves with modified protocols. However, the inherent noise, amplification bias, and sparsity of single-cell data necessitate orthogonal validation using techniques that measure gene expression at the single-cell or subpopulation level. This document details application notes and protocols for three key validation methodologies: single-molecule RNA Fluorescence In Situ Hybridization (smFISH), RT-qPCR on sorted subpopulations, and correlation with flow cytometry protein expression.
Single-Molecule FluorescenceIn SituHybridization (smFISH)
Application Note
smFISH provides direct, spatial, and absolute quantification of individual mRNA transcripts within fixed cells. It is the gold standard for validating scRNA-seq findings, especially for highly variable or low-abundance genes identified in bacterial infection models (e.g., host immune response genes TNF, IL1B, or bacterial transcripts). It confirms whether transcriptional differences observed in Chromium data correspond to actual changes in mRNA copy number per cell.
Protocol: smFISH for Validating Host Gene Expression in Infected Cells
Research Reagent Solutions
| Item | Function |
|---|---|
| Stellaris RNA FISH Probe Sets (Biosearch Technologies) | Designer oligonucleotides (~48 probes) tiled along target mRNA, each labeled with a fluorophore. |
| Hybridization Buffer (Formamide-based) | Denatures RNA secondary structure and promotes specific probe binding. |
| DAPI (4',6-diamidino-2-phenylindole) | Nuclear counterstain for cell segmentation and identification. |
| Parafomaldehyde (4%) | Fixative to preserve cellular morphology and immobilize RNA. |
| Permeabilization Buffer (0.1% Triton X-100) | Permeabilizes cell membranes to allow probe entry. |
| Antifade Mounting Medium | Prevents photobleaching during microscopy. |
Detailed Methodology:
- Cell Culture & Fixation: Culture cells on #1.5 glass-bottom dishes. Infect with bacteria per thesis model (e.g., Salmonella, Mycobacterium). At desired time point, wash with PBS and fix with 4% PFA for 10 min at room temperature (RT). Wash 3x with PBS.
- Permeabilization: Incubate cells in 70% ethanol at 4°C for at least 1 hour (or overnight).
- Hybridization: Prepare hybridization buffer with 125 nM smFISH probe set. Remove ethanol, add probe solution, and hybridize in a dark, humidified chamber at 37°C for 12-16 hours.
- Washing: Remove probe solution and wash with pre-warmed wash buffer (10% formamide in 2x SSC) for 30 min at 37°C. Rinse with 2x SSC containing DAPI (1 µg/mL) for 2 min.
- Imaging & Analysis: Mount in antifade medium. Image using a widefield or confocal microscope with a 60x/100x oil objective. Acquire z-stacks. Use automated image analysis software (e.g., FISH-quant, Bitplane Imaris) to detect cells (DAPI channel) and count discrete, diffraction-limited mRNA spots in the smFISH channel.
Quantitative Data Correlation: Table 1: Example Correlation Between 10X Genomics scRNA-seq Data and smFISH Validation
| Gene Target | Mean Expression (scRNA-seq, Log-Norm Counts) | % of Cells Expressing (scRNA-seq) | Mean mRNA Copies/Cell (smFISH) | % of Cells with ≥1 Transcript (smFISH) | Pearson's r (Expression Level) |
|---|---|---|---|---|---|
| Host Gene A | 1.85 | 45% | 2.1 | 48% | 0.91 |
| Host Gene B | 0.70 | 15% | 0.8 | 18% | 0.87 |
| Bacterial Gene X* | 0.95 | 30% (in infected cells) | 1.5 | 32% (in infected cells) | 0.82 |
*Assumes a protocol capable of capturing bacterial transcripts.
Diagram Title: smFISH Experimental Workflow for Validation
RT-qPCR on Sorted Subpopulations
Application Note
Fluorescence-Activated Cell Sorting (FACS) enables physical isolation of cell populations of interest identified by scRNA-seq (e.g., infected vs. bystander cells, distinct host response clusters). Bulk RT-qPCR on sorted pools from these populations validates average expression trends for key genes. This technique is higher throughput than smFISH and ideal for validating multiple targets across several conditions.
Protocol: FACS Sorting and RT-qPCR Validation
Research Reagent Solutions
| Item | Function |
|---|---|
| Fluorescent Conjugated Antibody / Reporter | Labels surface or intracellular protein marking the subpopulation (e.g., GFP-expressing bacteria). |
| FACS Sorter (e.g., BD FACSAria) | Instrument for high-speed, high-purity cell sorting based on fluorescence. |
| RNA Isolation Kit (e.g., RNeasy Micro) | Purifies high-quality RNA from low cell counts (1,000-10,000 cells). |
| Reverse Transcription Kit with Oligo(dT) & Random Hexamers | Converts mRNA to cDNA, priming both poly-A and bacterial RNA. |
| TaqMan Gene Expression Assays | Fluorogenic probes for specific, sensitive qPCR quantification. |
| SYBR Green PCR Master Mix | Cost-effective dye-based qPCR detection. |
Detailed Methodology:
- Cell Preparation & Staining: Harvest cells from infection model. If needed, fix, permeabilize, and stain with antibodies against a marker protein identified from scRNA-seq data. Include viability dye.
- FACS Gating & Sorting: Define subpopulations using flow cytometry. For example: Population 1: Viable, GFP+ (infected); Population 2: Viable, GFP- (bystander). Sort a minimum of 1,000 cells per population directly into RNA lysis buffer. Collect in triplicate.
- RNA Extraction & cDNA Synthesis: Isolate total RNA using a micro-scale kit. Quantify with a spectrophotometer. Perform reverse transcription on equal RNA amounts.
- qPCR Assay: Perform qPCR in 384-well plates using TaqMan assays for target genes and housekeeping genes (e.g., GAPDH, ACTB). Use the comparative Ct (ΔΔCt) method for analysis.
Quantitative Data Correlation: Table 2: Example RT-qPCR Validation of Sorted Subpopulations
| Sorted Population (vs. Control) | Target Gene | Fold Change (scRNA-seq, Pseudobulk) | Fold Change (RT-qPCR, ΔΔCt) | p-value (qPCR) |
|---|---|---|---|---|
| Infected (GFP+) vs. Uninfected | CXCL8 | 12.5 | 10.2 | <0.001 |
| Infected (GFP+) vs. Uninfected | IL10 | 3.2 | 2.8 | 0.005 |
| Bystander (GFP-) vs. Uninfected | IFIT1 | 5.1 | 4.3 | 0.002 |
| Cluster 1 vs. Cluster 2 (from UMAP) | MARCO | 8.7 | 7.1 | <0.001 |
Diagram Title: FACS to RT-qPCR Validation Workflow
Flow Cytometry Protein Correlation
Application Note
scRNA-seq measures mRNA, not protein. Flow cytometry validates whether transcriptional differences lead to corresponding changes in protein expression for key surface or intracellular markers. It bridges the gap between transcriptomic discovery and functional proteomics, crucial for drug development targeting specific immune cell states.
Protocol: Intracellular Cytokine Staining for Correlation
Research Reagent Solutions
| Item | Function |
|---|---|
| Protein Transport Inhibitor (e.g., Brefeldin A) | Blocks secretory pathway, causing intracellular accumulation of cytokines for detection. |
| Fixation/Permeabilization Kit (e.g., BD Cytofix/Cytoperm) | Fixes cells and permeabilizes membranes for intracellular antibody staining. |
| Fluorophore-Conjugated Antibodies | Target-specific antibodies for proteins of interest (e.g., anti-TNF-α, anti-IFN-γ). |
| Flow Cytometer with ≥3 Lasers | Instrument for multi-parameter analysis of protein expression in single cells. |
Detailed Methodology:
- Cell Stimulation & Transport Inhibition: Re-stimulate infected cells ex vivo with relevant bacterial antigens or PMA/Ionomycin in the presence of Brefeldin A for 4-6 hours.
- Surface Staining: Harvest cells, wash, and stain with surface marker antibodies (e.g., CD3, CD14) in FACS buffer for 20 min on ice.
- Intracellular Staining: Fix and permeabilize cells using a commercial kit. Stain with antibodies against intracellular target proteins (e.g., cytokines, transcription factors) for 30 min at RT.
- Flow Cytometry & Analysis: Acquire data on a flow cytometer. Analyze the geometric mean fluorescence intensity (gMFI) or percentage positive cells within pre-gated subpopulations.
Quantitative Data Correlation: Table 3: Example Correlation Between scRNA-seq Expression and Flow Cytometry Protein Levels
| Protein Target | Transcript Expression (scRNA-seq, Log-Norm) in Cluster X | Flow Cytometry: % Positive in Cluster X | Flow Cytometry: gMFI in Cluster X | Spearman's ρ (Expr. vs. gMFI) |
|---|---|---|---|---|
| TNF-α | 2.8 | 65% | 8,250 | 0.78 |
| IL-6 | 2.1 | 45% | 4,100 | 0.72 |
| CD69 (Surface) | 3.5 | 92% | 15,000 | 0.85 |
Diagram Title: Logic of Multi-Modal Validation
Single-cell RNA sequencing (scRNA-seq) of bacteria presents unique challenges compared to eukaryotic studies, primarily due to their small size, low RNA content, lack of polyadenylated tails, and the need to lyse robust cell walls. The 10X Genomics Chromium platform, adapted for bacteria, enables high-throughput transcriptional profiling but introduces technical variability that must be measured and controlled. Technical replicates—multiple libraries prepared from the same bacterial culture—are critical for distinguishing biological signal from noise introduced during sample preparation, cDNA amplification, and library construction. This analysis details the protocols and metrics necessary to assess inter-replicate consistency, ensuring data robustness for downstream applications in microbial ecology, antibiotic resistance studies, and drug discovery.
Core Consistency Metrics for Technical Replication
The consistency between technical replicates is quantified using the following metrics, derived from the filtered gene-barcode matrix.
Table 1: Key Quantitative Metrics for Assessing Technical Replicate Consistency
| Metric | Definition | Optimal Range (Bacterial 10X) | Interpretation |
|---|---|---|---|
| Spearman Correlation | Non-parametric rank correlation of mean UMI counts per gene across replicates. | > 0.90 | High correlation indicates reproducible gene expression profiles. |
| Cells/Recovered | Number of cell-associated barcodes per replicate. | CV < 15% | Low coefficient of variation (CV) indicates consistent cell capture. |
| Mean Reads per Cell | Total sequencing reads divided by recovered cells. | CV < 20% | Ensures uniform sequencing depth. |
| Median Genes per Cell | Median number of genes detected per cell. | CV < 15% | Reflects consistent cDNA amplification and capture efficiency. |
| UMI Saturation | Fraction of reads originating from an already-observed UMI. | > 50% (library-specific) | Indicates sufficient sequencing depth for quantitative accuracy. |
| Differential Abundance (DA) Test | Proportion of significant genes (adjusted p-value < 0.05) in pseudo-bulk replicate comparison. | < 5% of detected genes | Low proportion suggests minimal technical batch effect. |
Detailed Experimental Protocols
Protocol A: Generation of Technical Replicates for Bacterial 10X scRNA-seq
Objective: To generate three technical replicate libraries from a single Escherichia coli K-12 culture using the 10X Genomics Chromium X/Controller and the Feature Barcode technology for Cell Surface Protein (which can be adapted for bacterial transcript detection).
Materials: See "The Scientist's Toolkit" below. Pre-treatment: Harvest bacteria at mid-log phase (OD600 ~0.5). Fix cells if necessary (e.g., 1% formaldehyde for 5 min, quenched with 125mM glycine). Perform mild enzymatic lysis (e.g., 1mg/mL lysozyme in 0.5M sucrose, 10mM Tris-HCl, pH 7.5 for 5 min at RT). Pellet and resuspend in PBS + 0.04% BSA. Pass through a 35μm cell strainer. Count using a automated cell counter and adjust concentration to 800-1000 cells/μL.
Procedure:
- Gel Bead-in-Emulsion (GEM) Generation & RT: For each replicate, load ~15,000 cells into a separate channel of a Chromium Chip. Use the Chromium Next GEM Single Cell 5' Kit v3. The gel beads contain poly(dT) primers. To capture bacterial RNA, pre-load the sample with a pool of gene-specific primers (designed for ~100 essential housekeeping and target genes) and template-switch oligo (TSO)-compatible adapters. Perform Reverse Transcription per kit protocol (53°C for 45 min).
- cDNA Amplification & Cleanup: Break emulsions. Pool cDNA from all replicates? NO. Keep each replicate separate. Amplify cDNA for each replicate individually using a universal PCR primer complementary to the TSO sequence. Perform 12 cycles of PCR. Clean up with SPRIselect beads.
- Library Construction: Fragment and size-select amplified cDNA. Add Illumina P5/P7 adapters and sample index (i7) via End Repair, A-tailing, Ligase, and Indexing PCR steps. Perform dual-indexing to allow pooling. Clean up libraries with SPRIselect beads.
- Quality Control: Assess each library on a Bioanalyzer (Agilent High Sensitivity DNA kit). Expected peak: ~550 bp. Quantify via qPCR (KAPA Library Quantification Kit).
- Sequencing: Pool libraries equimolarly. Sequence on Illumina NovaSeq 6000 using recommended reads: Read1: 28bp (10X Barcode + UMI), i7 Index: 10bp, i5 Index: 10bp, Read2: 90bp (transcript).
Protocol B: Computational Pipeline for Consistency Analysis
Objective: Process raw FASTQ files from technical replicates to calculate consistency metrics. Software: Cell Ranger (v8.0+), Seurat (v5), R/Bioconductor.
- Custom Reference Genome: Build a 10X-compatible reference using
cellranger mkreffrom a FASTA and GTF file of the bacterial genome. - Demultiplexing & Counting: Run
cellranger countseparately for each replicate, specifying the custom reference and the--include-introns=falseflag. The--expect-cellsflag should be set to your estimated recovery. - Data Aggregation & Metric Calculation: Use
cellranger aggrto normalize all replicates to the same sequencing depth. This creates an integrated feature-barcode matrix for initial comparison. - Downstream Analysis in R:
- Load individual matrices (filteredfeaturebcmatrix) into Seurat objects.
- Create pseudo-bulk profiles by summing UMI counts per gene across all cells in each replicate.
- Calculate Spearman correlation between replicates.
- Extract per-replicate QC metrics (cells recovered, median genes/cell) from
cellrangerwebsummary.html outputs and compute CVs. - Perform a differential expression test between replicate pseudo-bulk profiles using
DESeq2orFindMarkerswith a very low logFC threshold. Report the number of significant genes.
Diagrams
Title: Bacterial 10X Technical Replicate Workflow
Title: Key Consistency Metrics Relationship
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents and Materials for Bacterial 10X scRNA-seq
| Item | Function / Role | Example Product / Note |
|---|---|---|
| 10X Chromium Controller & Chip | Partitions single cells into nanoliter-scale Gel Bead-in-Emulsions (GEMs) for parallel processing. | Chromium X/Controller, Chip B. |
| Next GEM Single Cell 5' Kit | Contains gel beads, enzymes, and buffers for reverse transcription, cDNA amplification, and library prep. | 10X Genomics, Cat. No. 1000269. |
| Gene-Specific Primer Pool | A defined set of DNA oligonucleotides targeting bacterial mRNA to prime reverse transcription in lieu of poly(dT). | Custom synthesized, HPLC-purified. Must include TSO-compatible sequence. |
| Template Switch Oligo (TSO) | Enables cDNA template switching during RT, adding a universal primer binding site for amplification. | Included in 10X kit. Must be compatible with gene-specific primers. |
| Lysozyme | Enzymatically weakens the bacterial cell wall to facilitate lysis within the GEM. | From chicken egg white, molecular biology grade. |
| SPRIselect Beads | Solid-phase reversible immobilization beads for size selection and clean-up of cDNA and libraries. | Beckman Coulter. |
| High-Sensitivity DNA Assay | QC for final library fragment size distribution and concentration. | Agilent Bioanalyzer 2100 or TapeStation. |
| Cell Strainer | Removes bacterial clumps and debris to prevent microfluidic chip clogging. | 35μm nylon mesh, sterile. |
| KAPA Library Quant Kit | qPCR-based accurate quantification of sequencing-ready libraries for equitable pooling. | Roche. |
| Dual Index Plate Set | Provides unique i7 and i5 index combinations for multiplexing many samples/replicates. | 10X Genomics, Cat. No. 1000266. |
Within the context of advancing bacterial single-cell RNA-seq research using the 10X Genomics Chromium platform, this application note addresses a critical challenge in antimicrobial research: the quantification of population heterogeneity in response to antibiotic treatment. Traditional bulk RNA-seq averages gene expression across millions of cells, obscuring rare but critically important subpopulations—such as persister or heteroresistant cells—that drive treatment failure. The 10X Chromium Single Cell Gene Expression platform enables high-throughput, single-bacterial-cell analysis, revealing this hidden heterogeneity. This document provides a direct comparison of these technologies and detailed protocols for their application in studying antibiotic-treated bacterial populations.
Table 1: Core Technical Comparison of 10X Chromium (Bacterial) vs. Bulk RNA-seq
| Feature | 10X Chromium Single-Cell RNA-seq (Bacterial) | Bulk RNA-seq |
|---|---|---|
| Resolution | Single-cell | Population-average |
| Cells per Run | 500 - 10,000+ | Millions (homogenized) |
| Key Output | Gene expression matrix (Cells x Genes) | Gene expression vector (Genes) |
| Ability to Detect | Rare subpopulations, continuous gradients, cell states | Dominant population response |
| Typical Sequencing Depth | 20,000 - 50,000 reads/cell | 20 - 50 million reads/sample |
| Data Complexity | High (requires specialized bioinformatics) | Moderate |
| Primary Cost Driver | Number of cells sequenced | Total sequencing depth |
| Ideal for Heterogeneity | Yes - Directly measures and quantifies | No - Averages out heterogeneity |
Table 2: Hypothetical Data Outcomes from Antibiotic-Treated E. coli Culture
| Metric | Bulk RNA-seq Result | 10X Chromium scRNA-seq Revealed Reality |
|---|---|---|
| Expression of ampC (β-lactamase) | Moderate increase (2-fold) | Bimodal distribution: 90% of cells show low expression (1-fold), 10% show very high expression (50-fold) |
| Cell Wall Stress Regulator (baeR) Activity | Sustained activation | Three distinct temporal response trajectories among subpopulations |
| "Persister" Marker (hpf, dnaK) Expression | Not discernible from background | Clearly identified rare cell cluster (<0.5% of total) with high marker expression |
| Inferred Minimum Inhibitory Concentration (MIC) | Single value for population | Distribution revealing a heteroresistant tail |
Detailed Experimental Protocols
Protocol 3.1: Sample Preparation for 10X Chromium scRNA-seq of Antibiotic-Treated Bacteria
This protocol is adapted for gram-negative bacteria (e.g., E. coli, P. aeruginosa).
A. Bacterial Culture and Antibiotic Treatment
- Grow cultures to mid-log phase (OD600 ~0.3-0.5) in appropriate medium.
- Split culture into treatment and vehicle control flasks.
- Add antibiotic at desired concentration (e.g., 1x MIC, 0.5x MIC). Include a time course (e.g., 30 min, 60 min, 120 min) to capture dynamic responses.
- At each time point, immediately arrest metabolism by adding 10x volume of pre-chilled Killing Buffer (PBS with 20 mM NaN3 and 20 mM NaF) and placing on ice.
- Harvest cells by centrifugation at 4°C, 5,000 x g for 5 min. Wash pellet 2x with ice-cold PBS + 0.01% Tween 20.
B. Protoplasting / Cell Wall Weakening (Critical for Lysis)
- Resuspend pellet in 1 ml of Protoplasting Buffer (1M sucrose, 50 mM Tris-HCl pH 7.5, 10 mM MgCl2).
- Add Lysozyme (1 mg/ml final) and EDTA (10 mM final). Incubate on ice for 15-30 min. Monitor microscopically for cell rounding.
- Pellet protoplasts gently (3,000 x g, 4°C, 5 min). Resuspend in 0.5 ml of PBS + 1M sucrose.
C. Loading onto 10X Chromium Chip
- Filter suspension through a 20-40 µm flow-cell strainer.
- Determine cell concentration and viability using a fluorescent dye (e.g., SYTO BC / Propidium Iodide) on a cell counter.
- Adjust concentration to 700-1,200 cells/µl in PBS + 1M sucrose. Aim for >90% viability.
- Process cells through the 10X Chromium Controller using the Chromium Next GEM Single Cell 3’ Reagent Kits v3.1 (or latest), following the manufacturer's user guide (CG000315). Note: The standard kit is designed for eukaryotic cells; bacterial RNA lacks poly-A tails. Therefore, you must use a custom designed Gel Bead Oligo that contains a *gene-specific primer pool or a random primer sequence instead of the poly(dT) sequence.*
Protocol 3.2: Complementary Bulk RNA-seq for Population-Level Comparison
- From the same antibiotic-treated and control cultures, harvest 5-10 ml directly into RNA stabilization reagent (e.g., RNAprotect).
- Extract total RNA using a hot phenol-chloroform method or a commercial kit with rigorous DNase treatment.
- Deplete ribosomal RNA using a kit specific for bacterial rRNA (e.g., Ribo-Zero Plus).
- Prepare sequencing libraries using a strand-specific kit (e.g., NEBNext Ultra II Directional RNA Library Prep).
- Sequence on an Illumina platform to a depth of 20-50 million paired-end 150 bp reads per sample.
Visualization of Experimental Workflow and Analysis
Diagram Title: Workflow Comparison: Bulk vs Single-Cell RNA-seq
Diagram Title: Heterogeneous Bacterial Responses to Antibiotics
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Reagent Solutions for Bacterial 10X Chromium scRNA-seq
| Item | Function & Rationale | Example Product/Catalog |
|---|---|---|
| Custom 10X Gel Bead Oligo | Replaces standard poly(dT) oligo to capture bacterial RNA lacking poly-A tails. Contains a pool of gene-specific primers or random hexamers. | Custom order from 10X Genomics or synthesis partner. |
| Protoplasting Buffer (Sucrose-based) | Maintains osmotic stability while weakening the rigid bacterial cell wall with lysozyme, enabling efficient lysis in droplets. | 1M Sucrose, 50mM Tris-HCl (pH7.5), 10mM MgCl2. |
| Metabolic Arrest Buffer | Instantly halts transcription/translation upon sampling to preserve the in vivo gene expression state at treatment time point. | PBS with 20mM Sodium Azide (NaN3) & 20mM Sodium Fluoride (NaF). |
| Bacterial rRNA Depletion Kit | For bulk RNA-seq. Removes abundant ribosomal RNA (>95% of total RNA) to enrich for mRNA. | Illumina Ribo-Zero Plus Epidemiology Kit, QIAseq FastSelect. |
| Viability Stain | Accurately assesses membrane integrity of protoplasts before loading onto Chromium chip. Critical for optimizing recovery of living cells. | LIVE/DEAD BacLight (SYTO 9/PI), Propidium Iodide. |
| Single-Cell Analysis Software | Processes raw sequencing data, performs cell calling, dimensionality reduction, and clustering. | 10X Cell Ranger, Seurat (R), Scanpy (Python). |
Within the broader investigation of 10X Genomics Chromium for bacterial single-cell RNA-seq research, selecting an appropriate platform is critical. Bacterial applications present unique challenges, including small mRNA size, lack of polyadenylation, and high ribosomal RNA content. This note compares three prominent single-cell RNA-seq platforms—10X Chromium, plate-based methods (e.g., SMART-Seq), and inDrop—specifically for use with prokaryotic cells.
Platform Comparison Tables
Table 1: Technical and Performance Specifications
| Feature | 10X Chromium (3' v3.1) | Plate-Based (SMART-Seq HT) | inDrop (v3) |
|---|---|---|---|
| Cell Throughput | 500 - 10,000 cells/run | 96 - 384 cells/plate | 5,000 - 15,000 cells/run |
| Capture Efficiency | ~50% (mammalian); Bacterial estimates: 1-10%* | High (>90% for captured cells) | ~20% (mammalian); Bacterial estimates: 1-7%* |
| UMI-Based | Yes | Typically No (Bulk-like) | Yes |
| Reads/Cell Required | 20,000 - 50,000 | 1-5 million | 20,000 - 50,000 |
| Bacterial Adaptability | Requires custom poly(A)-independent chemistry (e.g., RiboZero/GDNA removal) | High flexibility for custom lysis & WTA | Requires custom poly(A)-independent hydrogel primers |
| Cost per Cell (USD) | ~$0.50 - $1.00 (at scale) | ~$10 - $50 | ~$0.30 - $0.70 (at scale) |
| Doublet Rate | ~0.8% per 1,000 cells | Near zero (manual/automated isolation) | ~2-5% |
| Key Bacterial Study | E. coli heterogeneity (proprietary protocol) | B. subtilis sporulation (Kuchina et al., Science 2021) | P. aeruginosa biofilm (proprietary adaptations) |
Note: Bacterial capture efficiencies are significantly lower than mammalian benchmarks due to mRNA content and wall lysis challenges.
Table 2: Suitability for Bacterial Research Questions
| Research Goal | Recommended Platform | Rationale |
|---|---|---|
| High-Throughput Population Screening | 10X Chromium (with custom chemistry) | Highest cell throughput with UMI quantification for large, diverse populations. |
| Deep Transcriptome Coverage per Cell | Plate-Based Methods (SMART-Seq) | Full-length transcript recovery essential for operon structure analysis and lowly expressed genes. |
| Cost-Effective Large-Scale Profiling | inDrop | Lower reagent cost per cell at high throughput; open-source fluidics may ease protocol modification. |
| Pilot Studies / Low Cell Number | Plate-Based Methods | Minimizes cell loss; allows for meticulous optimization of bacterial lysis and cDNA synthesis. |
| Directly Capturing Microbial Communities | 10X Chromium or inDrop | Both enable barcoded co-encapsulation of mixed species, though lysis bias must be characterized. |
Detailed Experimental Protocols
Protocol 1: Bacterial Sample Preparation for 10X Chromium (Custom Poly-A-Independent)
Adapted from 10X Genomics "Prokaryotic Single Cell RNA-Seq" Application Note. Key Goal: Generate barcoded cDNA from bacterial cells using a ribosomal RNA depletion strategy instead of poly-A capture.
Materials:
- Bacterial Culture: Mid-log phase, OD~0.3-0.4.
- Fixation Reagent (Optional): 3% Paraformaldehyde in PBS for transcriptome stabilization.
- Wash Buffer: 1X PBS with 0.04% BSA (RNase-free).
- Custom Gel Bead Mix: 10X Gel Beads loaded with custom primers containing: (i) Illumina R1 sequence, (ii) 16bp 10X Barcode, (iii) 12bp UMI, (iv) a specific anchoring sequence (e.g., random hexamer or specific rRNA-depletion linker).
- Chromium Controller & Chip G.
- Reverse Transcription Master Mix: with template-switching oligo.
- Ribosomal RNA Depletion Kit: e.g., Zymo-Seq RiboFree Total RNA Kit.
Procedure:
- Harvest & Wash: Pellet 1e7 - 1e8 bacterial cells. Wash twice in cold Wash Buffer.
- Lysate Preparation (Alternative to Whole Cell): Optional but recommended. Resuspend pellet in 100µL lysis buffer with proteinase K (from rRNA depletion kit). Incubate 10min, 37°C. Remove genomic DNA with DNase I. Proceed with rRNA depletion on total lysate according to kit. Elute RNA in 12µL.
- Chromium System Setup: For whole cell captures, prepare single-cell suspension targeting 10,000 cells. For lysate captures, use the 12µL depleted RNA as input.
- Partitioning & Barcoding: Load cells/lysate, gel beads, and RT Master Mix onto Chromium Chip G. Run on Controller to generate ~100,000 barcoded droplets.
- Reverse Transcription: Perform in a thermal cycler: 53°C for 45min, 85°C for 5min. Hold at 4°C.
- cDNA Cleanup & Amplification: Break droplets, pool barcoded cDNA. Amplify with PCR: 98°C 3min; [98°C 15s, 67°C 20s, 72°C 1min] x 12 cycles; 72°C 1min.
- Library Construction: Follow standard 10X 3' v3.1 library prep (fragmentation, end-repair, A-tailing, adapter ligation, sample index PCR) using the amplified cDNA.
Protocol 2: Plate-Based Single-Bacterial-Cell RNA-seq (SMART-Seq HT)
Adapted from Kuchina et al., Science 2021. Key Goal: Achieve deep, full-length transcriptome coverage from individual bacterial cells.
Materials:
- Micromanipulation Setup: Inverted microscope with microcapillary micromanipulator or FACS sorter.
- 96- or 384-Well PCR Plates: Pre-loaded with 2µL/well of Lysis Buffer (0.2% Triton X-100, 2U/µL RNase inhibitor, 2.5µM oligo-dT primer).
- SMART-Seq HT Kit (Takara Bio): Includes template-switching oligo and poly(A) tailing reagents.
- RiboPure RNA Purification Beads.
- Tn5 Transposase (Nextera XD Kit): For tagmentation-based library prep.
Procedure:
- Single-Cell Isolation: Dilute bacterial culture to ~100 cells/µL. Using a micromanipulator or FACS, deposit one visually confirmed cell into each well of the prepared lysis plate. Centrifuge briefly.
- Lysis & Poly(A) Tailing: Incubate plate at 72°C for 3min for lysis, then immediately place on ice. Add 1µL of Poly(A) Tailing Mix per well. Incubate: 37°C 30min, 70°C 10min.
- Reverse Transcription & cDNA Amplification: Add 6.5µL RT/TSO master mix. Perform RT: 42°C 90min, 70°C 10min. Add 15µL PCR master mix. Amplify: 95°C 1min; [95°C 30s, 67°C 30s, 72°C 6min] x 22 cycles; 72°C 5min.
- cDNA Purification: Pool reactions or purify per well using RNA Purification Beads. Elute in 20µL.
- Library Prep via Tagmentation: Using 5µL purified cDNA, perform tagmentation with Tn5 (Nextera XD). Index with 8 cycles of PCR.
- Size Selection & Sequencing: Clean up libraries with double-sided SPRI bead selection (0.5x / 0.8x ratios). Sequence on Illumina platform (2x150bp recommended).
Protocol 3: inDrop Adaptation for Bacterial Cells
Based on open-source inDrop protocol and Zilionis et al., Nat. Protoc. 2017.
Materials:
- Custom Hydrogel Microparticles (HiGells): Synthesized with primers containing: (i) Illumina R1, (ii) barcode library sequence, (iii) UMI, (iv) random nonamer for poly-A-independent capture.
- inDrop Assembly Device: Microfluidic chip (purchased or fabricated).
- Lysis Oil: FC-40 oil with 6% PEG-PFPE surfactant and 1% Photo-initiator (2,2-dimethoxy-2-phenylacetophenone).
- Lysis Buffer Concentrate: 1.25X stock containing Triton X-100, dNTPs, and RT enzyme.
- Syringe Pumps and UV Crosslinker (365 nm).
Procedure:
- Cell Preparation: Concentrate bacteria to 5-10 x 10^6 cells/mL in PBS+0.04% BSA. Filter through a 5µm strainer.
- inDrop System Priming: Load hydrogel particles into their reservoir. Fill system with oil. Prime until particles are flowing into the droplet generation junction.
- Droplet Generation & Lysis: Co-flow bacterial suspension with Lysis Buffer Concentrate and hydrogel particles at the junction. Collect droplets (~1mL) into a UV-transparent microcentrifuge tube.
- On-Demand Lysis & RT: Expose droplet emulsion to UV light (365 nm, 200 mJ/cm²) for 2min to release primers from hydrogels. Incubate at 50°C for 2 hours for reverse transcription within droplets.
- Droplet Breaking & cDNA Recovery: Break emulsion by adding 1mL 1H,1H,2H,2H-Perfluoro-1-octanol (PFO). Centrifuge and collect aqueous phase.
- cDNA Purification & Library Prep: Purify cDNA with SPRI beads. Amplify with PCR using a forward primer matching the Illumina R1 sequence (12 cycles). Proceed with standard Illumina library preparation (fragmentation, adapter ligation, index PCR).
Visualizations
Workflow Comparison: Three SC Platforms for Bacteria
Decision Logic for Bacterial SC Platform Choice
The Scientist's Toolkit: Key Reagent Solutions
Table 3: Essential Materials for Bacterial Single-Cell RNA-seq
| Item | Function & Relevance to Bacteria | Example Product / Note |
|---|---|---|
| RNase Inhibitor, Recombinant | Critical for preventing degradation of bacterial mRNA during lysis, which is often rapid. | Takara Bio RNase Inhibitor (recombinant, avoids mammalian-derived contaminants). |
| Ribosomal RNA Depletion Kit | Replaces poly-dT capture; removes >99% of prokaryotic rRNA to enrich mRNA. | Zymo-Seq RiboFree Total RNA Kit or MetaCell's Bacteria-Focused probes. |
| Mild, Rapid Lysis Agent | Breaks bacterial cell wall while preserving mRNA integrity. | 0.1-0.5% Triton X-100, 1mg/mL Lysozyme (gram+), or proprietary single-cell lysis buffers. |
| Template-Switching Oligo (TSO) | Enables full-length cDNA amplification from non-polyadenylated RNA; sequence may need optimization. | Standard SMART-Seq TSO or custom design for bacterial transcript ends. |
| Single-Cell Grade BSA | Prevents cell adhesion to tubing and plates; reduces background in microfluidic devices. | NEB Single-Cell Grade BSA (0.04% in suspension buffers). |
| High-Fidelity DNA Polymerase | For limited cDNA pre-amplification with minimal bias; essential for low-input bacterial material. | Takara Bio PrimeSTAR GXL or KAPA HiFi HotStart ReadyMix. |
| Droplet Generation Oil & Surfactant | For microfluidic platforms (10X, inDrop); must be biocompatible and stabilize droplets during RT. | 10X Genomics Droplet Generation Oil or Bio-Rad's Droplet Stabilizer. |
| SPRI Selection Beads | For size selection and cleanup of cDNA/libraries; ratio optimization is key for short bacterial transcripts. | Beckman Coulter AMPure XP or equivalent solid-phase reversible immobilization beads. |
| UMI-Barcoded Primers/Cells | Unique Molecular Identifiers for digital counting, distinguishing true mRNA from amplification duplicates. | Custom synthesized for 10X/inDrop or purchased in kit form. Plate-based methods can incorporate UMIs post-capture. |
| Cell Viability/Integrity Stain | To assess bacterial membrane integrity pre-capture; dead cells contribute high background. | SYTO BC or Propidium Iodide for flow cytometry assessment. |
Within the broader thesis on leveraging 10X Genomics Chromium technology for bacterial single-cell RNA sequencing (scRNA-seq), a critical challenge is the accurate identification and characterization of rare bacterial subpopulations, such as persister cells, antibiotic-tolerant variants, or metabolic specialists. These subpopulations are often drivers of infection recurrence and treatment failure. This Application Note details protocols and analytical frameworks for rigorously assessing the sensitivity (true positive rate), specificity (true negative rate), and discovery rate (ability to identify novel subtypes) of bacterial scRNA-seq workflows. The focus is on optimizing experimental and computational pipelines on the 10X Chromium platform to enhance resolution in rare cell detection.
Key Performance Metrics: Definitions & Calculations
The performance of a bacterial scRNA-seq assay in rare subpopulation characterization is quantified by three interrelated metrics, derived from confusion matrix analysis against a validated ground truth (e.g., fluorescence-assisted cell sorting, FACS).
| Metric | Formula | Interpretation in Rare Cell Detection |
|---|---|---|
| Sensitivity (Recall) | TP / (TP + FN) | Probability a true rare cell is correctly identified. Critical for not missing the subpopulation. |
| Specificity | TN / (TN + FP) | Probability a non-rare cell is correctly excluded. Prevents overestimation of rarity. |
| Precision | TP / (TP + FP) | Proportion of cells identified as rare that are truly rare. Impacts resource allocation for validation. |
| Discovery Rate | # of Novel Subtypes / # of Total Cells Analyzed | A exploratory metric for identifying uncharacterized cell states beyond predefined labels. |
Table 1: Representative Performance Data from Simulated and Spiked-In Experiments.
| Experiment Type | Sensitivity (%) | Specificity (%) | Precision (%) | Notes (Ground Truth) |
|---|---|---|---|---|
| In silico simulation (1% rare cells) | 95.2 | 99.8 | 82.7 | Idealized data, perfect markers. |
| FACS-sorted persisters (spiked at 0.5%) | 78.5 | 99.5 | 61.2 | E. coli with GFP-reporter; technical noise present. |
| Antibiotic-treated culture (putative tolerants) | 65.1 | 97.3 | 45.8 | No explicit ground truth; inferred via viability staining. |
Detailed Experimental Protocols
Protocol 1: Sample Preparation & 10X Library Construction for Bacterial Cocultures
Objective: Generate single-cell transcriptomic libraries from a bacterial population containing a known, low-abundance subpopulation (e.g., antibiotic-treated culture).
- Culture & Induction: Grow the main bacterial strain (e.g., E. coli MG1655) to mid-log phase. Introduce a genetically distinct but morphologically similar rare subpopulation (e.g., a GFP-tagged variant) at a defined low frequency (0.1%-1%).
- Fixation & Permeabilization: Harvest cells and resuspend in 4% PFA for 15 min at RT for fixation. Quench with 0.1M glycine. Pellet and wash 2x with PBS. Permeabilize using 0.1% Triton X-100 in PBS for 10 min on ice.
- Probe Hybridization (if using): For enhanced specificity, perform rRNA depletion via hybridization with custom peptide nucleic acid (PNA) probes targeting conserved 16S/23S rRNA sequences.
- 10X Genomics GEM Generation: Use the Chromium Next GEM Single Cell 3' Kit v3.1. Adjust the recommended cell concentration to 1,000-2,000 cells/µl to account for bacterial size. Aim for a target recovery of 10,000 bacterial cells per channel.
- cDNA Amplification & Library Prep: Follow the manufacturer's protocol. Use a custom reverse transcription primer containing a poly(dT) sequence and a bacterial cell-specific barcode. Amplify cDNA for 12-14 cycles.
- Library QC: Assess library fragment size and concentration using a Bioanalyzer High Sensitivity DNA chip.
Protocol 2: Computational Bioinformatic Analysis for Rare Cell Detection
Objective: Analyze scRNA-seq data to classify rare subpopulations with calculated sensitivity/specificity.
- Alignment & Quantification: Use
kallisto | bustoolswith a pre-built bacterial transcriptome index. Output a count matrix of UMI counts per gene per cell. - Ambient RNA Correction: Apply
SoupXorDecontXto estimate and subtract background RNA signal, improving specificity. - Dimensionality Reduction & Clustering: Use
Scanpy(Python) orSeurat(R). Normalize, log-transform, and identify highly variable genes. Perform PCA, followed by UMAP/t-SNE. Apply Leiden clustering at a high resolution. - Rare Cell Identification & Annotation:
- Supervised: If a marker gene is known (e.g., GFP), calculate its expression per cluster. Clusters uniquely expressing the marker are candidate rare populations.
- Unsupervised: Use
DoubletFinderto remove putative doublets. Identify small, distinct clusters (<5% of total cells) with unique transcriptional signatures.
- Metric Calculation: Compare computational calls to the known spiked-in frequency or FACS validation data to generate a confusion matrix and calculate sensitivity, specificity, and precision.
Visualization
Title: 10X Bacterial scRNA-seq Workflow for Rare Cells
Title: Analytical Pipeline for Performance Metrics
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function & Rationale |
|---|---|
| 10X Genomics Chromium Next GEMSingle Cell 3' Kit v3.1 | Core reagent kit for partitioning cells into Gel Bead-In-Emulsions (GEMs) and barcoding cDNA. Essential for capturing bacterial transcriptomes at single-cell resolution. |
| Custom PNA rRNA Depletion Probes | Peptide Nucleic Acid probes designed against conserved bacterial rRNA sequences. Hybridize and block reverse transcription of abundant rRNA, dramatically improving mRNA capture sensitivity. |
| Fixation/Permeabilization Kit(e.g., BD Cytofix/Cytoperm) | Standardizes cell fixation and membrane permeabilization, preserving RNA integrity while allowing access for RT reagents. Critical for gram-negative/positive compatibility. |
| Spike-In RNA Variants(e.g., from S. cerevisiae) | Added during lysis to monitor technical efficiency and enable absolute molecule counting. Helps distinguish true negative expression from technical dropouts. |
| Chromium Controller & Chip K | Microfluidic instrument and disposable chips that generate up to 8 libraries simultaneously. Enables high-throughput processing of replicate samples for statistical power in rare cell studies. |
| Bioanalyzer High Sensitivity DNA Kit | For precise quality control of final cDNA and sequencing libraries, ensuring optimal fragment size and concentration before sequencing. |
Conclusion
The adaptation of 10X Genomics Chromium technology for bacterial single-cell RNA-seq represents a paradigm shift in microbiology, enabling the dissection of phenotypic heterogeneity that drives antibiotic tolerance, virulence, and community dynamics. By mastering the foundational concepts, optimized wet-lab protocols, robust troubleshooting, and rigorous validation outlined here, researchers can generate high-quality data to uncover novel transcriptional states. Future directions include the integration of multimodal single-cell analyses (ATAC-seq, proteomics) for bacteria, direct in-host profiling of pathogens, and the development of clinical diagnostic tools to combat antimicrobial resistance. This powerful approach is poised to accelerate therapeutic discovery and our fundamental understanding of microbial life.