Unveiling the Microbial World: A Comprehensive Guide to DNA Extraction for Metagenomic Analysis
This article provides a detailed roadmap for researchers, scientists, and drug development professionals navigating the critical first step in metagenomic studies: DNA extraction.
Unveiling the Microbial World: A Comprehensive Guide to DNA Extraction for Metagenomic Analysis
Abstract
This article provides a detailed roadmap for researchers, scientists, and drug development professionals navigating the critical first step in metagenomic studies: DNA extraction. We explore the foundational principles and sample-specific challenges that inform method selection. A comprehensive review of current commercial kits, phenol-chloroform, and mechanical lysis protocols is presented, followed by targeted troubleshooting strategies for common pitfalls like low yield, shearing, and inhibitor contamination. Finally, we delve into validation frameworks, benchmark studies, and comparative analyses to guide the selection of the optimal extraction strategy for specific research goals, from biomarker discovery to functional gene screening. This guide synthesizes the latest methodologies to ensure the integrity of your metagenomic data from sample to sequence.
The Foundation of Metagenomics: Why DNA Extraction is the Most Critical Step
Within a thesis on DNA extraction methods for metagenomic samples research, defining high-quality DNA is paramount. High-quality metagenomic DNA is the foundational material that determines the success of downstream applications like shotgun sequencing, qPCR, and functional gene array analysis. It must be characterized by high molecular weight, high purity, sufficient quantity, and faithful representation of the original microbial community structure without biases introduced during extraction.
Key Quality Metrics
High-quality metagenomic DNA is assessed against the following quantitative benchmarks:
Table 1: Quantitative Metrics for High-Quality Metagenomic DNA
| Metric | Target Value/Range | Assessment Method | Importance |
|---|---|---|---|
| Concentration | > 5 ng/µL (for direct sequencing) | Fluorometry (e.g., Qubit) | Ensures sufficient material for library prep; avoids PCR inhibitors. |
| Purity (A260/A280) | 1.8 - 2.0 | Spectrophotometry (e.g., Nanodrop) | Ratios outside range indicate protein (low) or RNA (high) contamination. |
| Purity (A260/A230) | > 2.0 | Spectrophotometry | Low values indicate contamination by humic acids, phenols, or salts. |
| Molecular Weight/Integrity | > 20 kb, smeared above 10 kb | Pulsed-Field or standard gel electrophoresis | High MW indicates minimal shearing, crucial for long-read sequencing and assembly. |
| Fragment Size Distribution | Majority > 1 kb | Fragment Analyzer, Bioanalyzer | Confirms integrity and suitability for library preparation protocols. |
| Inhibitor Presence | PCR amplification of a control gene (e.g., 16S rRNA) | PCR followed by gel electrophoresis | Confirms DNA is amplifiable and free of enzymatic inhibitors. |
| Community Representativity | Matches expected profile from sample type | qPCR of taxonomic markers, spike-in controls | Ensures extraction did not disproportionately lyse certain taxa. |
Technical Support Center
Troubleshooting Guides & FAQs
Q1: My extracted DNA has a low A260/A280 ratio (<1.8). What does this mean and how can I fix it? A: A low A260/A280 ratio typically indicates protein contamination (e.g., from inefficient protease K digestion or incomplete separation during phase separation).
- Solution: Repurify the DNA using a phenol:chloroform:isoamyl alcohol (25:24:1) extraction followed by ethanol precipitation. Alternatively, use a commercial cleanup kit designed for protein removal. Ensure adequate incubation time and temperature with protease during lysis.
Q2: My DNA yield is consistently low from soil samples. How can I improve it? A: Low yield often stems from inefficient cell lysis or DNA binding/retention on inhibitors.
- Solution: Implement a more rigorous mechanical lysis step (e.g., bead beating for 3-5 minutes at high speed). Pre-treat samples with agents to dissolve humics or use a lysis buffer containing CTAB or SDS to disrupt complex matrices. Include a known amount of internal standard (spike-in DNA) to quantify loss.
Q3: The DNA appears degraded on the gel (smear < 1 kb). What caused this and how do I prevent it? A: Degradation is caused by endogenous nucleases or excessive physical shearing.
- Solution: Ensure samples are flash-frozen immediately after collection and stored at -80°C. Include nuclease inhibitors during lysis. Avoid vortexing or pipetting lysates vigorously after cell disruption. Process samples on ice and use wide-bore tips during handling.
Q4: Downstream PCR consistently fails, even with good spectrophotometric readings. Why? A: This is a classic sign of co-extracted enzymatic inhibitors (humic acids, polysaccharides, polyphenols) not detected by A260/A230.
- Solution: Spectrophotometry is not sensitive to all inhibitors. Use fluorometry for accurate concentration. Dilute the DNA template (inhibitors dilute out faster than DNA). Perform a DNA cleanup using a kit with inhibitor-removal technology (e.g., based on silica membranes with inhibitor wash buffers). Validate with a PCR amplification of a control gene.
Q5: How do I know if my extraction is biased and not representing the true microbial community? A: Bias can arise from differential lysis of Gram-positive vs. Gram-negative cells, or selective loss of DNA.
- Solution: Use a standardized mock community with known composition to validate your extraction protocol. Incorporate a combination of enzymatic (lysozyme), chemical (SDS), and mechanical (bead beating) lysis steps to maximize diversity. Compare results from multiple extraction kits/methods on the same sample.
Experimental Protocol: Standardized Phenol-Chloroform Extraction with Bead Beating for Soil
Objective: To extract high-molecular-weight, inhibitor-free metagenomic DNA from complex soil samples.
Materials:
- Lysis Buffer (500 mM NaCl, 50 mM Tris-HCl pH 8.0, 50 mM EDTA, 4% SDS)
- Phenol:Chloroform:Isoamyl Alcohol (25:24:1)
- Chloroform:Isoamyl Alcohol (24:1)
- Isopropanol and 70% Ethanol
- TE Buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0)
- Bead-beating tubes with 0.1 mm silica/zirconia beads
- Proteinase K (20 mg/mL)
- Lysozyme (50 mg/mL)
Method:
- Weigh 0.25 g of soil into a bead-beating tube.
- Add 500 µL of lysis buffer, 50 µL of lysozyme, and 50 µL of proteinase K. Mix gently.
- Incubate at 37°C for 30 minutes with horizontal shaking.
- Add 0.5 g of beating beads. Secure tube and process in a bead beater at maximum speed for 2 minutes.
- Centrifuge at 10,000 x g for 5 minutes at 4°C to pellet soil debris and beads.
- Transfer supernatant to a new 2 mL tube.
- Add an equal volume of Phenol:Chloroform:Isoamyl Alcohol. Vortex vigorously for 30 seconds. Centrifuge at 12,000 x g for 10 minutes at room temperature.
- Transfer the upper aqueous phase to a new tube.
- Add an equal volume of Chloroform:Isoamyl Alcohol. Vortex and centrifuge as in step 7.
- Transfer the aqueous phase to a new tube. Add 0.7 volumes of room-temperature isopropanol. Mix by inversion and incubate at room temp for 10 minutes.
- Centrifuge at 15,000 x g for 30 minutes at 4°C to pellet DNA.
- Carefully decant supernatant. Wash pellet with 1 mL of 70% ethanol. Centrifuge at 15,000 x g for 10 minutes.
- Air-dry pellet for 10-15 minutes. Resuspend in 50-100 µL of TE Buffer. Store at -20°C or -80°C.
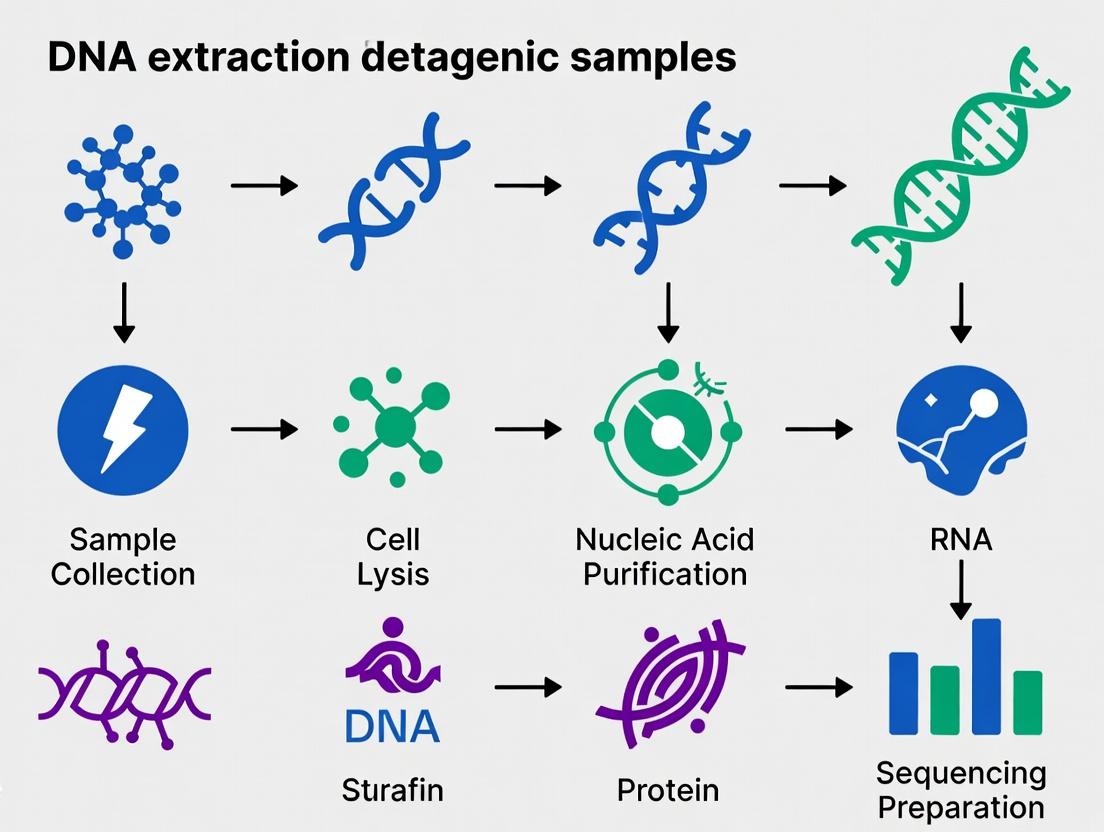
Visualization: Workflow for Assessing DNA Quality
Diagram Title: Quality Control Workflow for Metagenomic DNA
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for High-Quality Metagenomic DNA Extraction
| Item | Function | Key Consideration |
|---|---|---|
| Inhibitor Removal Technology Buffers | Specifically formulated to bind and remove humic acids, polyphenols, and polysaccharides during purification. | Critical for difficult samples (soil, sediment, manure). Look for proprietary resins or wash buffers. |
| Mechanical Lysis Beads (0.1mm) | Provides physical shearing force to break tough cell walls (e.g., Gram-positive bacteria, spores). | Material (zirconia/silica) and size are crucial for lysis efficiency and minimizing DNA shearing. |
| Phase Separation Reagents (Phenol:Chloroform:IAA) | Effectively denatures and removes proteins from the DNA-containing aqueous phase. | Requires careful handling; chloroform step removes residual phenol. |
| High-Salt Binding Buffers | Promotes efficient binding of large DNA fragments to silica membranes in spin columns. | Essential for recovering high-molecular-weight DNA and avoiding small fragment bias. |
| Nuclease-Free Water & TE Buffer | Final resuspension of DNA eluate. Maintains pH and chelates Mg2+ to prevent degradation. | Always use certified nuclease-free reagents to prevent degradation of your sample. |
| Internal Standard (Spike-in DNA) | Non-native DNA added at lysis start to quantitatively track extraction efficiency and bias. | Allows for absolute quantification and identifies sample-specific loss. |
Troubleshooting Guides & FAQs
Q1: Why is my extracted metagenomic DNA yield from soil samples consistently low, and how can I improve it? A: Low yield is often due to inefficient cell lysis or DNA adsorption to soil particles (e.g., humic acids). Current best practices involve:
- Protocol Adjustment: Use a combination of mechanical (e.g., bead beating for 45-60 seconds) and chemical lysis (e.g., CTAB buffer with proteinase K).
- Inhibit Co-precipitation: Increase the temperature of the lysis buffer to 70°C and incorporate a pre-wash step with a buffer like 120 mM sodium phosphate (pH 8.0) to desorb cells from particles.
- Inhibition Removal: Use specialized commercial kits with enhanced humic acid removal columns. Data from a 2023 comparative study is summarized below.
Q2: How do I choose between a commercial kit and a manual phenol-chloroform protocol for my specific sample type? A: The choice balances bias, yield, and inhibitor removal. See the comparative table below for guidance based on recent meta-analyses.
Q3: My sequencing results show an overrepresentation of Gram-negative bacteria. What step likely introduced this bias? A: This is a classic lysis bias. Gram-positive bacteria have thicker peptidoglycan layers and are harder to lyse.
- Solution: Optimize the lysis intensity. For bead beating, test a range of times (30s to 90s) and use a mixture of bead sizes (e.g., 0.1mm and 0.5mm). Validate with a mock microbial community of known composition. Avoid relying solely on enzymatic lysis.
Q4: What are the best practices for storing different sample types (swab, soil, water) prior to DNA extraction to minimize community shift? A: Immediate freezing at -80°C is ideal. If not possible:
- Soil/Sediment: Store at -20°C for short periods. For longer storage, use commercial preservation buffers (e.g., RNAlater) but validate for your sample, as they can introduce bias.
- Water: Filter onto a membrane (e.g., 0.22µm polyethersulfone) and place the filter in preservation buffer or freeze immediately.
- Swabs: Place the swab in a sterile tube with a stabilization buffer and freeze.
Q5: How can I verify that my extraction protocol is not introducing significant bias? A: Incorporate a standardized mock microbial community (comprising known ratios of diverse cells) into your experimental workflow. Extract DNA from this mock community alongside your samples and sequence it. Deviations from the expected taxonomic profile indicate protocol-induced bias.
Summarized Quantitative Data
Table 1: Comparison of DNA Extraction Methods for Diverse Sample Types (2023-2024 Meta-Analysis)
| Method / Kit (Example) | Avg. Yield (ng DNA/g soil) | Shannon Index Bias (vs. Gold Standard) | Humic Acid Removal (A260/A230 Ratio) | Best For Sample Type |
|---|---|---|---|---|
| PowerSoil Pro Kit | 18.5 ± 6.2 | Low (-0.15 ± 0.08) | High (2.1 ± 0.3) | Soil, Sediment, Feces |
| FastDNA SPIN Kit | 25.3 ± 9.1 | Moderate (-0.32 ± 0.11) | Moderate (1.8 ± 0.4) | Microbial Cultures, Biofilms |
| Phenol-Chloroform-IAA | 30.1 ± 12.5 | High (-0.51 ± 0.15) | Low (1.2 ± 0.5) | Water, Low-Biomass Filters |
| Modified CTAB Protocol | 22.4 ± 7.8 | Low (-0.19 ± 0.09) | High (2.0 ± 0.3) | Plant-Rhizosphere, High-Humic Soil |
Table 2: Impact of Bead Beating Duration on Lysis Efficiency and DNA Fragmentation
| Bead Beating Time (s) | Relative Yield (Gram+) | Relative Yield (Gram-) | Mean Fragment Size (bp) |
|---|---|---|---|
| 30 | 1.0 (Baseline) | 2.5 ± 0.3 | 12,000 ± 1,500 |
| 60 | 2.8 ± 0.4 | 2.7 ± 0.2 | 8,500 ± 1,200 |
| 90 | 3.1 ± 0.3 | 2.5 ± 0.3 | 5,200 ± 900 |
| 120 | 3.0 ± 0.5 | 2.1 ± 0.4 | 3,800 ± 750 |
Detailed Experimental Protocols
Protocol 1: Modified CTAB-Based Extraction for High-Humic Acid Soils
- Pre-wash: Weigh 0.5 g of soil. Add 1 ml of 120 mM sodium phosphate buffer (pH 8.0). Vortex for 5 minutes. Centrifuge at 10,000 x g for 2 min. Discard supernatant.
- Lysis: Add 750 µl of pre-heated (70°C) CTAB Lysis Buffer (2% CTAB, 1.4 M NaCl, 100 mM Tris-HCl pH 8.0, 20 mM EDTA) and 50 µl of Proteinase K (20 mg/ml). Vortex.
- Mechanical Disruption: Add ~0.3 g of a 1:1 mix of 0.1mm and 0.5mm zirconia/silica beads. Bead beat at 6.5 m/s for 60 seconds.
- Incubation: Incubate at 70°C for 30 minutes with gentle inversion every 10 minutes.
- Separation: Centrifuge at 12,000 x g for 5 min. Transfer supernatant to a new tube.
- Cleanup: Add 1 volume of phenol:chloroform:isoamyl alcohol (25:24:1). Mix thoroughly. Centrifuge at 12,000 x g for 10 min. Transfer aqueous phase.
- Precipitation: Add 0.7 volumes of isopropanol and 0.1 volumes of 3M sodium acetate (pH 5.2). Incubate at -20°C for 1 hour. Pellet DNA at 15,000 x g for 20 min.
- Wash & Elute: Wash pellet with 70% ethanol. Air-dry and resuspend in 50 µl TE buffer or nuclease-free water.
Protocol 2: Validation Using a Mock Microbial Community
- Standard: Use a commercially available, sequenced mock community (e.g., ZymoBIOMICS Microbial Community Standard).
- Spike-In: In parallel with your experimental samples, process an aliquot of the mock community (per manufacturer's instructions) through your entire extraction and library preparation protocol.
- Sequencing & Analysis: Sequence the mock community extract on the same run as your samples. Analyze the resulting taxonomic profile using the manufacturer's expected ratios as a reference.
- Bias Calculation: Calculate relative log2 fold-change for each member. A protocol with minimal bias will show small deviations across all members.
Diagrams
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function & Rationale |
|---|---|
| Zirconia/Silica Beads (0.1mm & 0.5mm mix) | Provides mechanical shearing for robust cell wall disruption, especially critical for Gram-positive bacteria and spores. A mix of sizes increases lysis efficiency across diverse cell types. |
| CTAB (Cetyltrimethylammonium bromide) | A cationic detergent effective in lysing cells and precipitating polysaccharides and humic acids, which are common PCR inhibitors in environmental samples. |
| Proteinase K | A broad-spectrum serine protease that degrades proteins and inactivates nucleases, crucial for improving yield and DNA integrity during lysis. |
| PCR Inhibition Removal Columns | Specialized silica-based columns with buffers optimized to bind DNA while allowing humic acids, polyphenols, and other common environmental inhibitors to pass through. |
| Mock Microbial Community Standard | A defined mix of microbial cells with known genomic sequences and ratios. Serves as an essential process control to quantify bias introduced by the extraction and sequencing workflow. |
| Polyvinylpolypyrrolidone (PVPP) | An additive used to bind and precipitate polyphenolic compounds, which are potent PCR inhibitors found in plant-derived and some soil samples. |
| RNAlater / DNA/RNA Shield | Commercial stabilization buffers that rapidly penetrate tissues to inhibit RNase/DNase activity and preserve microbial community composition at the moment of sampling. |
Technical Support Center
Troubleshooting Guides & FAQs
Q1: My DNA yield from soil samples is consistently low. What are the primary culprits and how do I address them? A: Low yield from soil is often due to humic acid co-purification inhibiting lysis or DNA binding. Key steps:
- Increase mechanical disruption: Use bead-beating with a mixture of zirconia/silica beads (0.1 mm and 0.5 mm) for 2-3 cycles of 45 seconds each, with 2-minute ice pauses.
- Implement stringent humic acid removal: Use polyvinylpolypyrrolidone (PVPP) or activated charcoal wash steps in your column-based protocol. See Table 1 for optimized buffer additives.
- Validate with a spiked control: Introduce a known quantity of Pseudomonas fluorescens cells prior to extraction to calculate extraction efficiency.
Q2: How can I reduce host DNA contamination in gut microbiome extractions? A: Host depletion is critical. Implement a differential lysis step:
- Gentle lysis: Resuspend sample in PBS + 1% saponin, incubate at 37°C for 30 min to lyse mammalian cells. Centrifuge (500 x g, 10 min) to pellet intact microbial cells.
- Wash pellet: Resuspend microbial pellet in a harsh lysis buffer (e.g., containing SDS and proteinase K) and proceed with bead-beating.
- Alternative: Use commercial host depletion kits (e.g., MolYsis) that selectively degrade mammalian DNA.
Q3: My water sample filters clog immediately, and I cannot process sufficient volume for low-biomass analysis. What should I do? A: For turbid water, pre-filtration is essential.
- Serial Filtration: Use a cascade of pre-filters (e.g., 5.0 µm pore size cellulose filter) before the final collection filter (typically 0.22 µm polyethersulfone).
- Alternative Concentration: For very large volumes (>10 L), consider tangential flow filtration (TFF) to concentrate biomass without clogging.
- Direct Capture: For clear oligotrophic water, use a Sterivex filter housing which is less prone to clogging and allows for in-situ lysis.
Q4: When extracting from extreme environment samples (e.g., high salinity, low pH), my standard kits fail. How do I modify my approach? A: These matrices require extensive pre-washing to remove PCR inhibitors.
- High Salinity (Hypersaline): Dialyze the sample or dilute with molecular-grade water to reduce salt concentration below 0.5 M before filtration or centrifugation. Use ethanol washes with higher percentages (80-90%) to precipitate DNA effectively.
- Low pH (Acidic): Neutralize the sample with a sterile, mild buffer (e.g., Tris-HCl pH 8.0) immediately upon collection to prevent acid-mediated DNA degradation. Increase the concentration of chelating agents (EDTA) in your lysis buffer.
Q5: My metagenomic library shows extreme bias toward Gram-negative bacteria. How can I improve lysis of Gram-positive organisms? A: This indicates incomplete lysis. Enhance your protocol:
- Add Lysozyme: Incubate sample with lysozyme (10-20 mg/mL) at 37°C for 30-60 minutes prior to bead-beating.
- Incorporate Mutanolysin: For tough peptidoglycan, add mutanolysin (5 U/mL) to the lysozyme step.
- Optimize Bead-beating: Ensure your bead-beating matrix includes 0.1 mm beads for maximum physical disruption. Do not exceed 5 minutes total beating time to avoid DNA shearing.
Table 1: Optimized Pre-Lysis Additives for Inhibitor Removal by Sample Type
| Sample Matrix | Primary Inhibitor(s) | Recommended Additive | Concentration | Incubation (Pre-Lysis) |
|---|---|---|---|---|
| Soil (High Organics) | Humic/Fulvic Acids | Polyvinylpolypyrrolidone (PVPP) | 5% (w/v) | 30 min, RT with rotation |
| Sediment | Clay, Heavy Metals | Sodium Phosphate Buffer (pH 8.0) | 100 mM | 10 min, RT, then centrifuge |
| Fecal/Gut | Bile Salts, Polysaccharides | Phosphate Buffered Saline (PBS) Washes | 1X | 3x serial washes, pellet |
| Water (Wastewater) | Heavy Metals, Polyphenols | Chelex 100 Resin | 5% (w/v) | 20 min, 55°C with vortex |
| Extreme (High Salt) | Various Salts | Molecular Grade Water Wash | N/A | Dialysis or 10x dilution |
Table 2: DNA Yield & Quality Benchmark by Extraction Method
| Method | Soil (ng/g) | Fecal (ng/mg) | Water (ng/L) | 260/280 | 260/230 | Avg. Fragment Size (bp) |
|---|---|---|---|---|---|---|
| PowerSoil Pro Kit | 45.2 ± 12.1 | 220.5 ± 45.3 | 15.8* | 1.85 ± 0.05 | 1.95 ± 0.10 | 15,000 |
| Phenol-Chloroform (w/ bead-beating) | 68.5 ± 25.7 | 310.8 ± 80.2 | 22.4* | 1.75 ± 0.10 | 0.80 ± 0.30 | 23,000 |
| Methanol-ENVA Lysis | 12.1 ± 3.5 | 95.6 ± 20.1 | 5.5* | 1.90 ± 0.03 | 2.05 ± 0.05 | 8,500 |
From 1L filtered, concentrated biomass. *Low 260/230 indicates phenol carryover.
Experimental Protocols
Protocol 1: Comprehensive Bead-Beating Lysis for Diverse Matrices
- Materials: Lysis Buffer (100 mM Tris-HCl pH 8.0, 100 mM EDTA pH 8.0, 1.5 M NaCl, 1% CTAB, 2% SDS), Bead Tube (0.1, 0.5 mm zirconia/silica mix), Proteinase K (20 mg/mL), β-mercaptoethanol.
- Method:
- Transfer up to 500 mg sample to bead tube.
- Add 750 µL lysis buffer and 20 µL proteinase K. Mix.
- Add 50 µL β-mercaptoethanol (for rich organic samples).
- Secure tubes and bead-beat at 6.0 m/s for 45 seconds.
- Incubate at 55°C for 15 minutes.
- Repeat steps 4 and 5 twice (3 cycles total).
- Centrifuge at 13,000 x g for 5 min at 4°C.
- Transfer supernatant to a clean tube for purification.
Protocol 2: Inhibitor Removal via PVPP Column Wash (for Soil/Sediment)
- Materials: Empty chromatography column, PVPP powder, Wash Buffer (5.0 M Guanidine HCl, 20 mM Tris-HCl pH 6.6).
- Method:
- Prepare a 2 mL column bed of hydrated and washed PVPP.
- Equilibrate column with 5 mL Wash Buffer.
- After initial lysate clearing (from Protocol 1, step 8), mix lysate with an equal volume of Wash Buffer.
- Load mixture onto PVPP column. Collect flow-through.
- Wash column with 2 mL Wash Buffer, combine with flow-through.
- Proceed with standard silica-column or SPRI bead-based DNA purification on the combined eluate.
Visualizations
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function | Key Consideration |
|---|---|---|
| Zirconia/Silica Beads (0.1 mm) | Maximizes physical cell disruption for tough Gram-positive bacteria and spores. | Can cause significant DNA shearing; optimize time. |
| Polyvinylpolypyrrolidone (PVPP) | Binds and removes polyphenolic compounds (humic acids) from environmental samples. | Must be pre-washed to remove contaminants. |
| Cetyltrimethylammonium Bromide (CTAB) | Ionic detergent effective for lysis and precipitating polysaccharides & humics in high-organic matrices. | Forms precipitate with salt; requires warm solutions. |
| Proteinase K (Recombinant, Lyophilized) | Broad-spectrum serine protease degrades cellular proteins and nucleases, protecting nucleic acids. | Quality is critical; ensure nuclease-free. |
| Guanidine Hydrochloride (GuHCl) | Chaotropic salt denatures proteins, inhibits nucleases, and promotes DNA binding to silica. | Competes with ethanol in binding buffers; concentration must be precise. |
| Sodium Phosphate Buffer (pH 8.0) | Competes with DNA for binding sites on clay particles in sediments, improving elution yield. | More effective than Tris-based buffers for clay-rich samples. |
| SPRI (Solid Phase Reversible Immobilization) Beads | Magnetic beads that bind DNA in PEG/High-Salt conditions for scalable, automatable purification. | Size selection is possible by adjusting PEG/salt concentration. |
| MolYsis-type Reagents | Selectively degrade mammalian cells/DNA in host-associated samples to enrich microbial DNA. | Effectiveness varies by sample type (e.g., saliva vs. stool). |
Troubleshooting Guides & FAQs
Q1: My DNA yield from soil metagenomic samples is consistently low after bead beating. What could be wrong? A: Low yield after mechanical lysis (bead beating) often indicates insufficient lysis of robust microbial cells (e.g., Gram-positive bacteria, spores) or DNA degradation. Troubleshooting steps:
- Verify Bead and Cycle Parameters: Increase the bead beating time (e.g., from 30s to 60-90s) or the number of cycles. Use a mixture of bead sizes (e.g., 0.1mm and 0.5mm) for more effective, heterogeneous cell disruption.
- Check for Overheating: Excessive beating generates heat, activating nucleases. Always perform beating in short, pulsed cycles with cooling intervals on ice.
- Pre-Treatment Consideration: For complex samples, incorporate a pre-treatment step with a mild chemical or enzymatic lysis (e.g., lysozyme incubation for 30 mins at 37°C) prior to bead beating to weaken cell walls.
Q2: I observe sheared/fragmented DNA when using sonication. How can I optimize for longer fragments suitable for long-read sequencing? A: Sonication is a high-shear mechanical method. To preserve high molecular weight (HMW) DNA:
- Optimize Energy Input: Use lower amplitude/power settings (e.g., 20-30% amplitude) and shorter total process time. Perform in multiple, very short pulses (e.g., 3-5 seconds ON, 15-30 seconds OFF on ice).
- Sample Viscosity: Ensure your sample is not too viscous. Dilute the lysate or increase lysis buffer volume to allow for more even energy distribution and cavitation.
- Protocol Shift: Consider switching to a gentler mechanical method like freeze-thaw cycling or a non-mechanical method if HMW DNA is critical.
Q3: When using SDS-based chemical lysis for gut microbiome samples, my protein contamination is high, inhibiting downstream enzymes. How do I resolve this? A: SDS is a strong ionic detergent that effectively lyses cells but co-solubilizes proteins.
- Increase Cleanup: Follow lysis with a more stringent protein precipitation step. Add ammonium acetate to a final concentration of 2M after lysis, incubate on ice for 10 minutes, then centrifuge to pellet proteins before DNA precipitation.
- Alternative Detergent: For sensitive downstream applications, consider switching to a milder, non-ionic detergent like Triton X-100 or N-Lauroylsarcosine, though lysis efficiency for tough cells may decrease.
- Optimized Precipitation: Use a selective binding matrix (e.g., silica membrane columns) optimized for removing anionic detergent carryover, following the lysis step directly.
Q4: Enzymatic lysis with lysozyme alone is ineffective for my environmental sample. What enzymatic cocktails are recommended for broad-spectrum lysis? A: Single enzymes have narrow specificity. Use sequential or combinatorial cocktails:
- Broad-Spectrum Cocktail: Incubate sample first with lysozyme (targets Gram-positive peptidoglycan), followed by proteinase K (digests proteins and degrades nucleases), in the presence of EDTA (chelates Mg2+, destabilizing membranes and inhibiting DNases).
- For Complex Matrices: Add enzymes like mutanolysin (for Streptococci) or lysostaphin (for Staphylococci). For fungal contaminants in samples, include chitinase.
- Always Combine: Enzymatic lysis is rarely used alone for metagenomics. It is most effective as a pre-treatment step (37°C for 30-60 min) prior to a mild chemical lysis step (adding detergents).
Q5: How do I choose the best lysis method for an unknown or highly diverse metagenomic sample? A: Employ a tiered or parallel strategy to maximize community representation.
- Hybrid Protocol: Start with a gentle enzymatic pretreatment (lysozyme, proteinase K), followed by a moderate chemical lysis (SDS or CTAB), and finish with a short, controlled mechanical disruption (e.g., 45s of bead beating). This combination attacks most cell types.
- Benchmarking: Perform a small-scale comparison of DNA yield and quality (via gel electrophoresis and fluorometry) from three sub-samples lysed with: a) Mechanical only, b) Chemical+Enzymatic, c) Hybrid. Use the most comprehensive method.
- Critical Control: Include an internal standard (e.g., a known quantity of an easy-to-lyse bacterium not found in your sample) to quantify and compare absolute lysis efficiency across methods.
Table 1: Quantitative Comparison of Cell Lysis Methods for Metagenomic DNA Extraction
| Method (Category) | Typical Efficiency (DNA Yield) | DNA Fragment Size | Processing Time | Cost per Sample | Key Advantages | Key Disadvantages |
|---|---|---|---|---|---|---|
| Bead Beating (Mech.) | High (90-99% for most cells) | Low to Medium (5-20 kb, can be sheared) | Low (1-5 min active) | Low | Broad-spectrum, rapid, scalable, high yield. | Heat generation, DNA shearing, noise, aerosol risk. |
| Sonication (Mech.) | Medium-High | Very Low (0.5-5 kb, highly sheared) | Low (1-10 min) | Medium | Very effective for tough cells, tunable. | Extensive shearing, high heat, requires optimization. |
| Detergent-Based (Chem.) | Medium (Varies by cell type) | Very High (>50 kb possible) | Medium (30-60 min) | Very Low | Gentle on DNA, simple, low cost. | Inefficient for robust cells, high protein/salt carryover. |
| Enzymatic | Low (Narrow spectrum) | Very High (>50 kb) | High (30 min to 2 hrs) | High | Extremely gentle, specific, minimal shear. | Slow, expensive, narrow target range, requires buffer control. |
| Hybrid (e.g., Enzymatic + Mech.) | Highest (Broad spectrum) | Medium-High (10-40 kb) | High (1-2 hrs) | Medium-High | Maximizes community representation, balanced output. | More complex protocol, longer hands-on time. |
Detailed Experimental Protocols
Protocol 1: Hybrid Lysis for Complex Soil Metagenomes (Adapted from the International Soil Metagenome Protocol)
- Objective: Extract high-yield, high-molecular-weight DNA representative of total microbial community.
- Reagents: Lysis Buffer (100 mM Tris-HCl pH 8.0, 100 mM EDTA pH 8.0, 1.5 M NaCl), Lysozyme (50 mg/mL), Proteinase K (20 mg/mL), 20% SDS, Phenol:Chloroform:Isoamyl Alcohol (25:24:1), Isopropanol, 70% Ethanol.
- Steps:
- Weigh 0.5g of soil. Add to a sterile 2mL tube containing a mixture of 0.1mm and 0.5mm silica/zirconia beads.
- Add 500μL of pre-warmed (37°C) Lysis Buffer, 50μL of Lysozyme solution. Vortex briefly. Incubate at 37°C for 30 minutes with gentle shaking.
- Add 30μL of Proteinase K and 60μL of 20% SDS. Mix by inversion.
- Incubate at 55°C for 60 minutes, with gentle inversion every 15 minutes.
- Mechanical Disruption: Secure tubes in a bead beater homogenizer. Process at maximum speed for 45 seconds. Immediately place on ice for 2 minutes.
- Centrifuge at 14,000 x g for 5 min at 4°C. Transfer supernatant to a new tube.
- Add an equal volume of Phenol:Chloroform:Isoamyl Alcohol. Vortex vigorously for 30s. Centrifuge at 14,000 x g for 10 min at 4°C.
- Carefully transfer the upper aqueous phase to a new tube. Add 0.7 volumes of room-temperature isopropanol. Mix by inversion. Incubate at RT for 10 min.
- Centrifuge at 14,000 x g for 15 min at 4°C to pellet DNA. Wash pellet with 1mL of 70% ethanol. Air-dry for 5-10 min.
- Resuspend in 50-100μL of TE buffer or nuclease-free water. Quantify via fluorometry.
Protocol 2: Gentle Chemical Lysis for Planktonic Microbial Biomass
- Objective: Extract ultra-pure, very high molecular weight DNA with minimal shearing for long-read sequencing.
- Reagents: TES Buffer (50 mM Tris-HCl pH 8.0, 50 mM EDTA pH 8.0, 20% Sucrose), N-Lauroylsarcosine (10% solution), RNase A (10 mg/mL).
- Steps:
- Pellet 10^9 - 10^10 microbial cells by gentle centrifugation (5,000 x g, 10 min, 4°C). Resuspend pellet thoroughly in 500μL of ice-cold TES Buffer.
- Add 50μL of a freshly prepared Lysozyme solution (10 mg/mL in TES). Incubate on ice for 30 minutes.
- Add 60μL of 10% N-Lauroylsarcosine. Mix by very gentle inversion. Incubate on ice for 10 minutes. The solution should become viscous.
- Add 5μL of RNase A. Incubate at 37°C for 15 minutes.
- Proceed with protein removal via dialysis (preferred for HMW DNA) or a very gentle chloroform extraction. Avoid vortexing; use wide-bore pipette tips for all transfers.
- Precipitate DNA using ethanol or isopropanol without vigorous mixing. Spool DNA using a glass rod if possible.
- Resuspend DNA slowly in a large volume (e.g., 200μL) of TE buffer overnight at 4°C.
Visualizations
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Metagenomic Cell Lysis
| Reagent | Category | Primary Function in Lysis | Key Consideration for Metagenomics |
|---|---|---|---|
| Zirconia/Silica Beads (0.1mm & 0.5mm mix) | Mechanical | Physical disruption of cell walls via high-speed shaking. | Bead composition affects DNA binding; size mixture increases lysis spectrum. |
| Sodium Dodecyl Sulfate (SDS) | Chemical (Ionic Detergent) | Dissolves lipid membranes & denatures proteins. | Very effective but inhibits downstream enzymes if not removed; can co-precipitate with DNA in cold. |
| N-Lauroylsarcosine | Chemical (Ionic Detergent) | Membrane solubilization, nuclease inhibition. | Milder than SDS, often preferred for HMW DNA extraction from sensitive cells. |
| Lysozyme | Enzymatic | Hydrolyzes β-(1,4) linkages in peptidoglycan of Gram-positive bacteria. | Efficiency is pH and buffer dependent (requires Tris-EDTA). Ineffective alone for many environmental microbes. |
| Proteinase K | Enzymatic | Broad-spectrum serine protease; digests proteins and inactivates nucleases. | Requires detergent (SDS) for full activity. Essential for removing contaminating enzymes. |
| Ethylenediaminetetraacetic Acid (EDTA) | Chemical (Chelator) | Chelates Mg2+ and Ca2+, destabilizing membranes and inhibiting metallonucleases. | A critical component of almost all lysis buffers to protect DNA during extraction. |
| Cetyltrimethylammonium bromide (CTAB) | Chemical (Detergent) | Precipitates polysaccharides and denatures proteins; effective in removing humic acids from soil. | Used in specific protocols for humic acid-rich samples (e.g., soil, compost). |
| Phenol:Chloroform:Isoamyl Alcohol | Chemical (Organic Solvent) | Denatures and partitions proteins/lipids into organic phase, leaving nucleic acids in aqueous phase. | Critical for purifying DNA from complex samples. Handle with care under a fume hood. |
Welcome to the Technical Support Center for Metagenomic DNA Extraction. This resource addresses common challenges related to co-extracted inhibitors that interfere with downstream molecular applications.
Troubleshooting Guides & FAQs
Q1: My downstream PCR amplification from soil DNA fails, even with high-yield extraction. What is the most likely cause and how can I confirm it? A: Humic acids are the most prevalent inhibitor in environmental samples. They absorb at wavelengths similar to nucleic acids (A230/A260), inhibiting polymerase activity. Confirm by spectrophotometry: a high A260/A230 ratio (<1.7) and brownish DNA pellet indicate contamination.
Q2: My extracted DNA is viscous and difficult to pipette, leading to inconsistent library prep yields. What should I do? A: Viscosity suggests co-precipitation of polysaccharides (e.g., from plant or microbial cell walls). Confirm via gel electrophoresis: DNA may appear as a high molecular weight smear. Increase mechanical lysis (bead beating) time to fragment polysaccharides and add a pre-treatment step with a polysaccharide-degrading enzyme (e.g., pectinase for plant-rich samples).
Q3: I suspect protein contamination is affecting my restriction enzyme digestion in clone library construction. How can I mitigate this? A: Residual proteins, including nucleases, can remain bound to DNA. Use a phenol:chloroform:isoamyl alcohol (25:24:1) step post-lysis to denature and partition proteins. Follow with a high-salt precipitation or silica-column wash using optimized buffers containing guanidine thiocyanate to remove protein residues.
Q4: What is the most effective single metric for assessing inhibitor presence before costly sequencing? A: Perform a qPCR inhibition assay. Compare the amplification efficiency (Cq values) of your sample spiked with a known quantity of exogenous control DNA (e.g., phage lambda DNA) against a clean control. A significant Cq delay (>2 cycles) indicates inhibition.
Table 1: Diagnostic Signatures and Quantification of Common Inhibitors
| Inhibitor | Primary Source | Spectrophotometric Signature (Nanodrop) | Gel Electrophoresis Clue | Functional Assay Impact |
|---|---|---|---|---|
| Humic Acids | Soil, Sediment, Compost | Low A260/A230 (<1.7), elevated A340 | None specific | PCR inhibition at >0.1 µg/µL |
| Polysaccharides | Plant tissue, Biofilms, Sludge | Low A260/A230, Viscous sample | Smear, impeded migration | Inhibits restriction enzymes, pipetting errors |
| Proteins | Cellular debris, Lysozyme, RNase A | Low A260/A280 (<1.8) | None specific | Binds DNA, inhibits enzymatic steps |
| Phenolics | Woody plants, Herbaceous matter | Brown color, low A260/A230 | Discolored gel lane | Oxidizes to quinones, degrades DNA |
Experimental Protocols
Protocol 1: Assessing Inhibitor Load via qPCR Spike-In Assay
- Prepare a 1:10 and 1:100 dilution of your extracted metagenomic DNA in nuclease-free water.
- Create a master mix for a standard qPCR assay targeting a conserved gene (e.g., 16S rRNA). Aliquot it into three sets.
- Set A (Control): Add only known copies of control template DNA.
- Set B (Sample): Add the same copies of control template DNA + 2 µL of your undiluted/diluted sample DNA.
- Run qPCR. Calculate ∆Cq = Cq(Set B) - Cq(Set A). A ∆Cq > 2.0 indicates significant inhibition in the sample.
Protocol 2: Polyvinylpolypyrrolidone (PVPP) Spin-Column Treatment for Humic Acid Removal
- Materials: Empty spin columns, 6% (w/v) PVPP suspension in extraction buffer, wash buffer (10 mM Tris-HCl, pH 8.0).
- Method:
- Load 500 µL of PVPP suspension into a spin column. Centrifuge at 5000 x g for 2 min to pack.
- Apply up to 200 µL of crude nucleic acid lysate (post-lysis, pre-precipitation) to the packed PVPP bed.
- Centrifuge at 5000 x g for 5 min. The PVPP will bind polyphenols and humics.
- Collect the flow-through and proceed with standard precipitation or binding to a silica column.
Visualizations
Co-extraction and Removal of Metagenomic Inhibitors
Inhibitor-Aware DNA Extraction Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Primary Function in Inhibition Management |
|---|---|
| CTAB (Cetyltrimethylammonium Bromide) | Precipitates polysaccharides and humic acids during lysis, separating them from nucleic acids. |
| PVPP (Polyvinylpolypyrrolidone) | Binds polyphenols and humic acids via hydrogen bonding, used in spin-column or batch formats. |
| Guanidine Thiocyanate (GuSCN) | Chaotropic salt in silica-binding buffers; denatures proteins and enhances inhibitor removal during washes. |
| SPRI Beads (Solid-Phase Reversible Immobilization) | Selective binding of DNA by size in PEG/NaCl, effectively removing small inhibitor molecules. |
| Pectinase & Cellulase | Enzymatic pre-treatment to degrade plant-derived polysaccharide matrices before cell lysis. |
| Sephadex G-200 | Gel filtration matrix for size-exclusion chromatography to separate DNA from smaller inhibitors. |
| HEPES Buffer (vs. Tris) | Used in lysis buffers for samples with acidic pH (e.g., peat) to maintain buffering capacity and prevent humic acid co-extraction. |
Technical Support Center: Troubleshooting Guide for Extraction Bias in Metagenomics
FAQs & Troubleshooting
Q1: My downstream alpha diversity metrics (e.g., Shannon Index) show unexpected shifts between sample groups. Could this be due to extraction bias? A: Yes. Inconsistent lysis of different cell wall types (e.g., Gram-positive vs. Gram-negative bacteria, spores) during extraction skews the observed taxonomic abundance. This is a primary source of extraction bias.
- Troubleshooting Protocol:
- Spike-in Control: Introduce a known quantity of cells with a dissimilar cell wall (e.g., Bacillus subtilis spores into a mostly Gram-negative sample) prior to extraction.
- Quantify Recovery: Use qPCR targeting a marker gene of the spike-in organism post-extraction.
- Calculate Bias: Compare the recovery efficiency between sample types. A significant difference (>50%) indicates severe extraction bias.
- Action: Normalize downstream sequence data using the spike-in recovery factors, or re-optimize the physical (bead-beating) and chemical (lysozyme) lysis steps.
Q2: My differential abundance analysis (e.g., DESeq2) highlights many significant taxa, but I suspect these are technical artifacts. How can I verify? A: Correlate the physicochemical properties of your samples with the purported differentially abundant taxa.
- Troubleshooting Protocol:
- Measure Co-variates: Record sample pH, viscosity, and humic acid content (A260/A230 ratio) for each sample.
- Statistical Correlation: Perform a PERMANOVA or Mantel test between these physicochemical matrices and your beta-diversity (Bray-Curtis) matrix.
- Interpretation: A strong, significant correlation (p < 0.05) suggests extraction efficiency, not biology, is driving community differences. You must include these co-variates in your statistical model or use an extraction method validated for such sample types.
Q3: I am getting low sequencing depth for host-associated samples (e.g., mouse stool), and the host DNA is overwhelming microbial signals. What is the solution? A: This is host DNA contamination bias. The extraction method must selectively lyse microbial cells while leaving host cells intact.
- Troubleshooting Protocol:
- Pre-treatment: Implement a pre-lysis step with a mild detergent (e.g., SDS) or enzyme (e.g., pancreatin) to degrade host cells/ debris, followed by centrifugation to pellet intact microbial cells.
- Selective Lysis: Use a kit specifically designed for host depletion.
- QC: Always run extracted DNA on a gel or Fragment Analyzer. A strong high-molecular-weight band indicates persistent host DNA.
- Quantify: Use a host-specific qPCR (e.g., targeting the mammalian GAPDH gene) to assess depletion efficiency. Aim for >99% host DNA reduction.
Q4: How does extraction bias specifically impact functional potential prediction (e.g., from PICRUSt2)? A: Extraction bias alters the genomic template pool. If taxa with unique functional genes (e.g., secondary metabolite clusters from Actinobacteria) are under-lysed, their functional potential will be absent from predictions.
- Troubleshooting Protocol:
- Benchmarking: Extract a mock community with known genomes using your standard protocol and an optimized, harsh lysis protocol (e.g., extended bead-beating).
- Sequence & Predict: Perform shotgun metagenomics (not 16S) on both extracts. Run functional prediction on both datasets (e.g., via HUMAnN3).
- Compare: Identify KEGG Orthology (KO) groups or enzyme classes missing from the standard protocol extract. These represent the "functional blind spots" introduced by your bias.
Data Presentation: Comparative Recovery Efficiencies
Table 1: Impact of Lysis Method on Taxonomic Recovery from a Defined Mock Community
| Lysis Method (Protocol Variation) | Gram-negative Recovery (E. coli) | Gram-positive Recovery (S. aureus) | Spore Recovery (B. subtilis) | Humic Acid Co-extraction (A260/A230) |
|---|---|---|---|---|
| Gentate (15s vortex) | 98% ± 5 | 40% ± 12 | <1% | Low (1.8-2.0) |
| Bead-beating (45s) | 95% ± 3 | 92% ± 4 | 15% ± 5 | Moderate (1.5-1.8) |
| Bead-beating + Lysozyme (30min) | 97% ± 2 | 95% ± 3 | 85% ± 7 | High (1.2-1.5) |
| Enzymatic Lysis Only | 90% ± 8 | 85% ± 6 | 10% ± 4 | Very Low (2.0-2.2) |
Table 2: Downstream Bioinformatics Impact of Uncorrected Extraction Bias
| Analysis Step | Metric | Result with Bias | Result with Bias-Corrected Data | Potential Misinterpretation Risk |
|---|---|---|---|---|
| Taxonomic Profiling | Relative Abundance of Gram+ Phyla (Firmicutes) | Artificially Low | Matches Expected Mock Community | False negative for key taxa |
| Diversity Analysis | Beta-diversity (PCoA Plot) | Clustering by Extraction Kit | Clustering by True Biological Group | Spurious group differences |
| Differential Abundance | DESeq2 p-value for Lactobacillus | p < 0.01 (False Positive) | p = 0.45 (Not Significant) | Incorrect biomarker identification |
| Functional Prediction | PICRUSt2 Pathway Completeness | Missing "Sporulation" pathway | "Sporulation" pathway detected | Erroneous metabolic network gaps |
Experimental Protocol: Comprehensive Bias Assessment
Protocol: Integrated Extraction Bias Audit for Metagenomic Workflows
Objective: To quantify and correct for cell lysis efficiency bias and co-extraction inhibitor bias in environmental samples.
Materials:
- Environmental samples (e.g., soil, stool)
- Internal Standard Spike-in Mix (e.g., log-phase Pseudomonas fluorescens [Gram-], Micrococcus luteus [Gram+], heat-killed Bacillus thuringiensis spores)
- DNA extraction kits (2+ for comparison)
- Bead-beater, centrifuge, thermomixer
- Qubit fluorometer, qPCR system, Fragment Analyzer
- PCR inhibitors detection kit (e.g., based on SPUD assay)
Method:
- Spike-in Addition: Aliquot sample. Add a precise, known quantity (e.g., 10^4 cells) of the Internal Standard Spike-in Mix to each aliquot immediately before extraction.
- Parallel Extraction: Perform DNA extraction on each aliquot using the different kits/protocols in parallel. Include a negative control (extraction blank).
- DNA QC: Measure total DNA yield (Qubit). Assess purity via A260/A280 and A260/A230 ratios. Profile fragment size distribution (Fragment Analyzer).
- Inhibitor Quantification: Perform the SPUD assay on a diluted aliquot of each extract to quantify PCR inhibition levels.
- Spike-in Recovery Quantification: Use qPCR with specific primers for each spike-in organism to calculate extraction recovery efficiency (%) for each cell type.
- Sequencing & Bioinformatic Correction: Sequence all extracts. In silico, subtract spike-in reads. Normalize sample counts using recovery efficiency factors (e.g., divide counts for taxa with similar cell wall properties by their corresponding recovery %).
Mandatory Visualizations
Title: Extraction Bias Origins and Downstream Impacts
Title: Troubleshooting Workflow for Suspected Extraction Bias
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Managing Extraction Bias
| Item | Function & Rationale |
|---|---|
| Defined Mock Community (e.g., ZymoBIOMICS) | Contains a mix of defined microorganisms with varying cell wall strengths. Serves as a process control to benchmark lysis efficiency and sequencing accuracy. |
| Internal Standard Spike-ins (e.g., Synthetic dsDNA, Alien PCR Controls) | Non-biological DNA spikes added pre-extraction to monitor total recovery. Biological spikes (e.g., Pseudomonas putida) monitor bias. |
| Inhibitor Removal Beads/Resins (e.g., PVPP, Sepharose) | Selectively bind humic acids, polyphenols, and other co-extracted inhibitors that reduce PCR and sequencing efficiency. |
| Host Depletion Kit (e.g., selective lysis buffers) | Contains optimized buffers to lyse microbial cells while leaving host cells (e.g., human, mouse) intact, enriching for microbial DNA. |
| Lysozyme & Mutanolysin Enzymes | Enzymes that specifically degrade peptidoglycan in Gram-positive bacterial cell walls, complementing mechanical lysis. |
| SPUD Assay Kit | A qPCR-based assay using a known template to detect and quantify the level of PCR inhibitors in a DNA extract. |
| Size-selection Magnetic Beads | Allow removal of very small (degraded) or very large (host) DNA fragments, improving library prep efficiency for metagenomics. |
From Theory to Bench: A Step-by-Step Guide to Modern Extraction Protocols
Technical Support Center: Troubleshooting Guides & FAQs
Frequently Asked Questions
Q1: I am consistently getting low DNA yield from difficult, humic acid-rich soil samples with the DNeasy PowerSoil Pro Kit. What can I do? A1: Low yields from humic-rich soils often stem from incomplete inhibitor removal. Ensure thorough vortexing with the PowerBead Tubes for the full 10 minutes. After adding Solution IRS, incubate on ice for 5 minutes before centrifugation to enhance precipitation of impurities. Increasing the sample input up to the kit's maximum (e.g., 2g) can also improve total yield.
Q2: When using the MagMAX Microbiome Ultra Kit for fecal samples, I observe carryover of PCR inhibitors. How can I optimize the wash steps? A2: Inhibitor carryover in magnetic bead protocols is frequently due to incomplete bead pelleting or residual ethanol. Ensure the magnetic separation time is at least 2 minutes for each wash. After the final ethanol wash, extend the air-dry time to 10 minutes at room temperature to ensure complete ethanol evaporation before elution. Using the recommended heated elution (50°C) improves inhibitor removal.
Q3: My ZymoBIOMICS DNA Miniprep Kit results show bacterial community bias in downstream 16S sequencing. Which step is most critical for bias reduction? A3: The mechanical lysis step is critical. Do not reduce the bead-beating time. Use the recommended Zymo BashingBead Lysis Tubes and homogenize for the full 5 minutes in a high-speed vortex adapter or bead beater. This ensures equal lysis efficiency for both Gram-positive and Gram-negative bacteria, minimizing bias.
Q4: For the MagMAX kit, my DNA eluate has low purity (260/230 < 1.8). What is the likely cause? A4: A low 260/230 ratio indicates carryover of organic compounds or salts. This is often from the binding/wash buffers. Verify that the provided Magnetic Bead Mix is fully resuspended before use. Ensure the supernatant is completely removed after the Proteinase K digestion step without disturbing the pellet. A final wash with 80% ethanol (freshly prepared) can improve 260/230 ratios.
Q5: I need to process high-volume liquid samples (e.g., >5 mL water) with the ZymoBIOMICS DNA Kit. What protocol modifications are supported? A5: For large volumes, you must first concentrate biomass. Centrifuge the sample at >12,000 x g for 15 minutes. Discard the supernatant and resuspend the pellet in up to 750 µL of PBS or water. Then proceed with the standard protocol from the lysis step. Do not exceed the maximum lysis tube volume.
Comparative Performance Data
Table 1: Kit Specifications and Performance Metrics
| Feature / Metric | Qiagen DNeasy PowerSoil Pro | Thermo Fisher MagMAX Microbiome Ultra | Zymo Research ZymoBIOMICS DNA Miniprep |
|---|---|---|---|
| Sample Input (Max) | 2 g soil / 250 mg stool | 250 mg stool / 5 mL liquid | 750 mg soil / 200 mg stool |
| Technology | Silica-membrane spin column | Magnetic bead purification | Spin column with BashingBead Lysis |
| Processing Time | ~60 minutes | ~45 minutes | ~50 minutes |
| Average Yield (Fecal Sample) | 5 - 15 µg | 4 - 12 µg | 3 - 10 µg |
| Purity (A260/A280) | 1.8 - 2.0 | 1.8 - 2.0 | 1.8 - 2.0 |
| Inhibitor Removal (Humic Acids) | Excellent | Very Good | Good |
| Bias Reduction (Gram+ vs. Gram-) | Good | Very Good | Excellent |
| Cost per Prep (approx.) | $8 - $10 | $9 - $12 | $7 - $9 |
Table 2: Common Issues and Recommended Solutions
| Problem | DNeasy PowerSoil | MagMAX Microbiome | ZymoBIOMICS |
|---|---|---|---|
| Low Yield | Increase bead-beating; Check IRS incubation. | Ensure bead resuspension; Check magnetic separation. | Verify bead beating time; Do not reduce lysis volume. |
| Inhibitor Carryover | Repeat wash step AW2; Ensure full air-dry. | Extend ethanol dry time; Use heated elution. | Add optional post-lysis inhibitor removal step. |
| Sheared DNA / Short Fragments | Gentle inversion after lysis; Avoid vortexing post-lysis. | Use wide-bore tips during transfer; Reduce mixing vigor. | Inherent bead-beating may shear; optimize time if needed. |
| Column Clogging | Do not overload sample; Pre-clear lysate by centrifugation. | Not applicable (magnetic beads). | Filter lysate through provided column before binding. |
| Low 260/230 Ratio | Ensure complete AW2 wash; Final elution with TE buffer. | Use fresh 80% ethanol final wash. | Ensure all ethanol is evaporated before elution. |
Detailed Experimental Protocol for Comparative Analysis
Protocol: Comparative Evaluation of Extraction Kits for Fecal Metagenomics
1. Sample Preparation:
- Homogenize fresh or frozen (-80°C) human fecal sample in anaerobic PBS.
- Aliquot 200 mg (±10 mg) of wet weight slurry into 3 separate sterile tubes.
- Include one aliquot of a defined mock microbial community (e.g., ZymoBIOMICS Microbial Community Standard) as an extraction control for each kit.
2. Parallel DNA Extraction:
- Process one aliquot with each kit (DNeasy PowerSoil Pro, MagMAX Microbiome Ultra, ZymoBIOMICS DNA Miniprep) strictly according to respective manufacturer protocols.
- Critical Step: Use the same bead-beating instrument (e.g., Vortex Genie 2 with horizontal adapter) and time (5 min) for all kits that include mechanical lysis to standardize lysis efficiency.
- Elute all final DNA in 50 µL of provided elution buffer or nuclease-free water (pre-heated to 55°C).
- Store eluted DNA at -20°C immediately.
3. Post-Extraction QC & Analysis:
- Quantification: Use fluorometric assay (e.g., Qubit dsDNA HS Assay) for accurate yield measurement.
- Purity: Measure absorbance ratios (260/280, 260/230) via microvolume spectrophotometry.
- Integrity: Analyze fragment size distribution using TapeStation or Bioanalyzer (Genomic DNA ScreenTape).
- Downstream Application: Perform 16S rRNA gene amplicon sequencing (V4 region) and shotgun metagenomic sequencing on all extracts. Use bioinformatic tools (QIIME 2, MetaPhlAn) to assess community composition, alpha/beta diversity, and bias against the known mock community.
Visualization of Workflow Comparison
Title: Comparative DNA Extraction Workflow from Three Kits
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Reagent Solutions for Metagenomic DNA Extraction
| Item | Function / Purpose | Typical Example / Note |
|---|---|---|
| PowerBead / BashingBead Tubes | Mechanically disrupt tough microbial cell walls (Gram-positive, spores) via vortexing or beating. | Ceramic/silica beads in a lysis buffer. Critical for unbiased lysis. |
| Inhibitor Removal Solution (IRS) | Chemically precipitate humic acids, phenolics, and other common environmental inhibitors. | Specific to soil/humic-rich samples. Incubation temperature is key. |
| Magnetic Bead Mix | Bind nucleic acids selectively in the presence of chaotropic salts; enable automated processing. | Paramagnetic particles. Full resuspension before use is vital. |
| Proteinase K | Digest proteins and degrade nucleases, facilitating lysis and protecting released DNA. | Often used in combination with lysis buffer for stool samples. |
| Binding/Wash Buffers | Create conditions for DNA adsorption to silica (columns or beads); wash away contaminants. | Contain chaotropic salts (e.g., guanidine HCl) and ethanol. |
| Nuclease-Free Elution Buffer | Release purified DNA from the silica matrix. Low-ionic-strength, slightly alkaline. | TE buffer or Tris-HCl (pH 8.0-8.5). Heated elution (50-55°C) increases yield. |
| Mock Microbial Community Standard | Control for extraction bias, lysis efficiency, and downstream sequencing accuracy. | Defined mix of known bacteria/fungi (e.g., from Zymo Research, ATCC). |
| Fluorometric DNA Quantification Kit | Accurately measure double-stranded DNA concentration without interference from RNA or salts. | Preferable to UV absorbance for metagenomic samples (e.g., Qubit). |
Technical Support Center
Troubleshooting Guides & FAQs
Q1: After phase separation, my aqueous (top) layer is cloudy or the interphase is thick and diffuse. What went wrong? A: This is commonly due to incomplete cell lysis or excessive cellular debris. Ensure your lysis buffer is appropriate for your sample type (e.g., add lysozyme for Gram-positive bacteria in soil metagenomic preps). Centrifuge the lysate more thoroughly before adding phenol-chloroform-isoamyl alcohol. If the problem persists, repeat the extraction starting with a smaller volume of the cloudy aqueous layer and a fresh equal volume of the extraction mixture.
Q2: I see no visible interphase after centrifugation. Is my DNA lost? A: A missing interphase often indicates inefficient initial lysis where no genomic material was released. Re-optimize your lysis step. For complex metagenomic samples, consider bead-beating or enzymatic lysis tailored to the community. Verify your protocol's effectiveness using a control sample with known DNA content.
Q3: My DNA yield is low, especially from low-biomass environmental samples. How can I improve it? A: For metagenomic samples, maximize yield by:
- Increasing starting material volume where possible.
- Adding a carrier (e.g., glycogen, linear polyacrylamide) during ethanol precipitation to aid nucleic acid pelleting. Note: Glycogen can interfere with some downstream enzymatic reactions.
- Performing a back-extraction: save the organic phase and interphase after the first extraction, add a fresh volume of TE buffer, mix, and re-centrifuge. Pool this new aqueous layer with the first.
Q4: My extracted DNA has low A260/A230 ratios (<1.8), indicating contamination. A: Low A260/A230 suggests carryover of organic compounds (phenol, guanidine) or salts. Ensure you are carefully removing the aqueous layer without disturbing the interphase. Increase the number of chloroform-only washes (e.g., two washes instead of one). During precipitation, wash the pellet thoroughly with 70% ethanol, and consider a second 70% ethanol wash. Allow the pellet to air-dry sufficiently to evaporate residual ethanol before resuspension.
Q5: The extracted DNA is sheared or of low molecular weight. A: This is a critical issue for metagenomic library construction. Avoid vigorous vortexing during mixing; invert tubes gently. Use wide-bore pipette tips when handling high-molecular-weight DNA after extraction. Consider using a milder lysis method if appropriate for your sample.
Q6: Is phenol-chloroform-isoamyl alcohol extraction suitable for all metagenomic samples? A: While effective for removing proteins and inhibitors, it may not be optimal for samples high in humic acids (e.g., certain soils). Sequential protocols combining PCI extraction with inhibitor-specific purification columns (e.g., based on CTAB or activated charcoal) are often necessary for such challenging samples.
Table 1: Comparison of DNA Extraction Efficiency from a Mock Soil Community
| Extraction Method | Average Yield (μg/g soil) | A260/A280 | A260/A230 | Average Fragment Size (bp) | % Host Contamination (if spiked) |
|---|---|---|---|---|---|
| PCI Extraction Only | 5.2 ± 0.8 | 1.82 ± 0.03 | 1.5 ± 0.2 | >20,000 | 0.1% |
| PCI + Silica Column Clean-up | 4.1 ± 0.5 | 1.85 ± 0.02 | 2.1 ± 0.1 | ~15,000 | 0.1% |
| Commercial Kit (Bead-beating) | 8.5 ± 1.2 | 1.88 ± 0.02 | 2.0 ± 0.1 | ~10,000 | 0.1% |
Table 2: Troubleshooting Common Issues and Impact on Yield/Purity
| Observed Problem | Likely Cause | Impact on Yield | Impact on Purity (A260/280) | Corrective Action |
|---|---|---|---|---|
| Cloudy Interphase | Incomplete Lysis, High Debris | Severe Decrease | Moderate Decrease | Optimize lysis; Increase centrifugation time/speed before extraction. |
| Low A260/A230 | Organic Solvent or Salt Carryover | Mild to No Impact | Severe Decrease | Increase chloroform wash steps; Improve ethanol pellet washing. |
| No Pellet After Precipitation | Very Low DNA, No Carrier | Total Loss | N/A | Use a nucleic acid carrier; Precipitate at -20°C overnight. |
| Viscous Organic Layer | Genomic DNA in Organic Phase | Severe Decrease | N/A | Avoid mixing too vigorously; Ensure correct salt/pH in aqueous phase. |
Detailed Experimental Protocol for Metagenomic DNA Extraction
Title: Phenol-Chloroform-Isoamyl Alcohol (PCI) Extraction for Inhibitor-Rich Metagenomic Samples
Principle: This method separates DNA from proteins and lipids based on differential solubility. In the presence of chaotropic salts and at a slightly acidic pH (7-8), phenol denatures and precipitates proteins, while chloroform removes lipids and facilitates phase separation. Isoamyl alcohol reduces foaming. DNA remains in the aqueous phase.
Reagents:
- Lysis Buffer (e.g., SDS-based with EDTA, pH 8.0)
- Phenol:Chloroform:Isoamyl Alcohol (25:24:1, v/v), pH 7.5-8.0
- Chloroform:Isoamyl Alcohol (24:1, v/v)
- 3M Sodium Acetate (NaOAc), pH 5.2
- Isopropanol (molecular biology grade)
- 70% Ethanol (molecular biology grade)
- TE Buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0) or Nuclease-Free Water
- Proteinase K (optional, for tough samples)
- Lysozyme (optional, for bacterial lysis)
- Glycogen (20 mg/mL, optional carrier)
Procedure:
- Cell Lysis: Suspend 0.5 g of soil/sediment sample in 1 mL of lysis buffer. Add Proteinase K (final conc. 100 μg/mL) and/or lysozyme (final conc. 10 mg/mL). Incubate at 55°C for 1-2 hours with gentle agitation.
- Debris Removal: Centrifuge at 12,000 x g for 10 minutes at 4°C. Transfer the supernatant to a new 2 mL microcentrifuge tube.
- PCI Extraction: Add an equal volume of Phenol:Chloroform:Isoamyl Alcohol (pH ~8). Mix gently by inversion for 5 minutes. Do not vortex. Centrifuge at 12,000 x g for 10 minutes at room temperature.
- Aqueous Phase Recovery: Carefully transfer the upper, clear aqueous phase to a new tube. Avoid the white interphase and the lower organic phase.
- Chloroform Wash (Clean-up): Add an equal volume of Chloroform:Isoamyl Alcohol (24:1). Mix gently by inversion for 2 minutes. Centrifuge at 12,000 x g for 5 minutes.
- DNA Precipitation: Transfer the aqueous phase to a new tube. Add 0.1 volumes of 3M NaOAc (pH 5.2) and 0.7 volumes of ice-cold isopropanol. Add 1 μL of glycogen carrier (optional). Mix gently and incubate at -20°C for 30 minutes to overnight.
- Pellet Washing: Centrifuge at >12,000 x g for 20 minutes at 4°C. Carefully discard the supernatant. Wash the DNA pellet with 1 mL of ice-cold 70% ethanol. Centrifuge again for 5 minutes. Carefully discard the ethanol.
- Resuspension: Air-dry the pellet for 5-10 minutes (do not over-dry). Resuspend the DNA in 50-100 μL of TE Buffer or nuclease-free water. Store at -20°C or -80°C.
Diagrams
PCI Extraction Workflow for Metagenomics
Troubleshooting Low Purity DNA
The Scientist's Toolkit: Key Reagent Solutions
Table 3: Essential Reagents for PCI DNA Extraction
| Reagent | Function & Critical Property | Metagenomic Application Note |
|---|---|---|
| Phenol:Chloroform:Isoamyl Alcohol (25:24:1) | Phenol denatures proteins. Chloroform removes lipids and helps separate phases. Isoamyl alcohol prevents foaming. Must be pH-balanced (~7.8-8.0) to keep DNA in the aqueous phase. | The gold-standard for protein removal. Essential for humic-acid rich samples prior to column clean-up. |
| Chloroform:Isoamyl Alcohol (24:1) | Removes trace phenol from the aqueous phase after the initial PCI extraction. Phenol inhibits downstream enzymes (e.g., polymerases, ligases). | A critical clean-up step. Often repeated twice for challenging environmental samples. |
| 3M Sodium Acetate (NaOAc), pH 5.2 | Provides monovalent cations (Na+) that neutralize the negative charge on DNA phosphate backbones, reducing solubility. The acidic pH favors precipitation. | Standard precipitating salt. For very dilute DNA, ammonium acetate can be used to avoid co-precipitating dNTPs or short primers. |
| Glycogen (Molecular Grade) | An inert, co-precipitating carrier. Visibly marks the pellet location and increases recovery efficiency of low-concentration nucleic acids. | Highly recommended for low-biomass metagenomic extractions (e.g., deep ocean, clean room). Ensure it is nuclease-free. |
| Proteinase K | A broad-spectrum serine protease that degrades proteins and inactivates nucleases. Critical for robust lysis of diverse organisms in a community. | Use at high concentrations (0.1-1 mg/mL) and incubate >1 hour for complex samples like stool or soil. |
| TE Buffer (pH 8.0) | Resuspension buffer. Tris maintains pH, EDTA chelates Mg2+ to inhibit DNases. Slightly alkaline pH aids DNA solubility and stability. | Preferable to water for long-term storage. For PCR-sensitive apps, use low-EDTA or nuclease-free water. |
Technical Support Center
Troubleshooting Guide
Q1: After bead beating, my sample shows significant heat generation, and downstream PCR fails. What went wrong? A: Excessive heat denatures proteins and nucleic acids. This is typically caused by over-processing. Follow this protocol: Use a 4°C chilled adapter or short, pulsed cycles (e.g., 30 seconds ON, 90 seconds OFF, repeated 5-6 times). Ensure your bead tube is kept on ice before and immediately after beating. Monitor lysate temperature; it should not exceed 25°C. For critical samples, perform bead beating inside a cold room.
Q2: My sonication efficiency for cell lysis is inconsistent between runs. How can I standardize it? A: Inconsistent sonication is often due to variable sample volume, probe placement, or cavitation efficiency. Use this standardized protocol: 1) Keep sample volume constant (±10%). Use a 1/8" microtip for volumes 0.2-1 mL. 2) Set the probe tip 1 cm from the tube bottom. 3) Use 10 pulses of 10 seconds ON, 20 seconds OFF at 40% amplitude on ice. 4) Perform a "shearing check" via gel electrophoresis (1% agarose) to confirm consistent fragment size distribution (target 300-500 bp for metagenomics). Calibrate power output annually.
Q3: Cryogenic grinding yields a fine powder, but my subsequent DNA yield is low. What is the optimal workflow? A: Low yield after cryo-grinding often stems from incomplete tissue homogenization or inefficient powder transfer. Optimal Protocol: 1) Pre-cool mortar, pestle, and sample in liquid N₂ for 5 minutes. 2) Grind 50-100 mg tissue in short, vigorous bursts until a fine, homogeneous powder forms (~2-3 minutes). 3. While still frozen, use a pre-cooled spatula to swiftly transfer the powder to a lysis buffer-containing tube. Do not let the powder thaw. 4. Immediately vortex or proceed to a secondary lysis step (e.g., enzymatic). Thawing before buffer addition causes degradation.
Q4: I need to lyse a mixed community with Gram-positive bacteria and fungal spores. Which mechanical method combination is best? A: A sequential approach is most effective. Use this protocol: 1) Primary Lysis (Bead Beating): Use a mix of 0.1 mm (for bacteria) and 0.5 mm (for spores) silica/zirconia beads. Process for 3 cycles of 45 sec ON, 2 min OFF on ice. 2) Secondary Lysis (Sonication): Subject the supernatant from step 1 to mild sonication (5 pulses of 5 sec ON, 10 sec OFF at 30% amplitude) to further disrupt stubborn spores and shear DNA to optimal length. This combination maximizes community representation.
Frequently Asked Questions (FAQs)
Q: What is the ideal bead material and size for soil metagenomic DNA extraction? A: Zirconia/silica beads (0.1 mm) are ideal for microbial cell wall disruption in soil. Larger beads (2-3 mm) aid in macroscopic soil particle disaggregation. A mixed bead size strategy often yields the highest DNA quality and quantity from complex matrices.
Q: Can I use sonication to lyse plant tissues directly? A: Not recommended. Plant tissues contain tough cellulose and polysaccharides that dampen sonic energy. Always perform cryogenic grinding first to pulverize the cell wall, then apply sonication to the resulting powder in lysis buffer for complete organelle disruption.
Q: How do I prevent cross-contamination during cryogenic grinding? A: Thoroughly clean the mortar and pestle with detergent, rinse with ethanol, and autoclave. Between samples, submerge tools in liquid N₂ to freeze off residual material, then clean again. Consider using disposable polyethylene bags and a sealed grinding apparatus for high-throughput, sensitive work.
Q: For bead beating, what is the optimal sample-to-bead slurry volume ratio? A: The sample (plus lysis buffer) volume should not exceed 1/3 of the tube's total capacity. The bead volume should be roughly equal to the sample buffer volume. For a 2 mL tube, use ~0.3 mL beads and 0.3-0.5 mL sample/buffer. This ensures sufficient kinetic energy transfer.
Table 1: Optimized Parameters for Bead Beating of Soil Samples
| Parameter | Recommended Setting | Effect on Lysis | Notes |
|---|---|---|---|
| Bead Size | 0.1 mm (zirconia) | High efficiency for bacterial cells | Combine with 2 mm beads for soil clumps. |
| Bead Fill Volume | 1/3 of tube volume | Optimal kinetic energy transfer | Too little reduces efficiency; too much limits movement. |
| Beating Time | 3 x 45 sec cycles | Balances lysis with heat generation | Critical for heat-sensitive communities. |
| Pause Time (Ice) | 2 min between cycles | Prevents overheating (>25°C) | Mandatory for intact DNA recovery. |
| Sample Buffer Volume | Equal to bead volume | Ensures proper slurry formation | Adjust based on sample absorption. |
Table 2: Comparison of Mechanical Lysis Methods for Different Sample Types
| Sample Type | Recommended Primary Method | Key Parameter | Avg. DNA Yield (ng/mg) | Avg. Fragment Size (bp) |
|---|---|---|---|---|
| Gram-negative Bacteria | Sonication | Amplitude: 30%, 5x 10s pulses | 150-200 | 500-1000 |
| Gram-positive Bacteria | Bead Beating | 0.1mm beads, 3x 60s cycles | 80-120 | 2000-10000 |
| Fungal Mycelia | Cryogenic Grinding + Bead Beating | Liq. N₂ grind, then 0.5mm beads | 50-80 | 1000-5000 |
| Plant Leaf | Cryogenic Grinding + Sonication | Liq. N₂ grind, then 30% amplitude | 40-60 | 300-500 (post-shear) |
| Soil Metagenome | Bead Beating (mixed beads) | 0.1mm & 2mm beads, 3x 45s cycles | 10-30* | Varies widely |
*Yield heavily dependent on soil organic content.
Experimental Protocols
Protocol 1: Integrated Lysis for Soil Metagenomics (Bead Beating & Sonication)
- Weigh 0.25 g of soil into a 2 mL screw-cap tube containing 0.3 mL of 0.1 mm and 0.1 mL of 2 mm zirconia beads.
- Add 750 µL of commercially available DNA/RNA Shield and 250 µL of lysis buffer (e.g., with SDS and proteinase K).
- Secure tubes in a pre-chilled (4°C) bead beater adapter.
- Process at 6.5 m/s for 3 cycles of 45 seconds, with 2-minute pauses on ice between cycles.
- Centrifuge at 12,000 x g for 1 minute at 4°C.
- Transfer 500 µL of supernatant to a new microcentrifuge tube on ice.
- Sonicate using a microtip at 30% amplitude for 3 pulses of 10 seconds ON, 20 seconds OFF to shear DNA to ~500 bp.
- Proceed with standard phenol-chloroform extraction and isopropanol precipitation.
Protocol 2: Cryogenic Grinding for Tough Plant Tissue
- Pre-cool a porcelain mortar, pestle, and metal spatula by filling the mortar with liquid nitrogen for 5 minutes.
- Submerge 100 mg of flash-frozen leaf tissue in liquid nitrogen in the mortar.
- Grind vigorously with the pestle until a fine, homogeneous powder forms (~2-3 minutes). Add liquid nitrogen as it evaporates to keep sample frozen.
- Using the pre-cooled spatula, quickly transfer the frozen powder to a tube containing 1 mL of pre-warmed (65°C) CTAB lysis buffer.
- Vortex immediately and thoroughly to disperse powder before it thaws.
- Incubate at 65°C for 30 minutes with occasional inversion.
- Continue with chloroform extraction and SPRI bead cleanup.
Diagrams
Mechanical Lysis Decision Pathway for Metagenomic Samples
Integrated Workflow for Soil DNA Extraction & Library Prep
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Mechanical Lysis |
|---|---|
| Zirconia/Silica Beads (0.1 mm) | Provides high-density, inert particles for efficient physical disruption of microbial cell walls during bead beating. |
| DNA/RNA Shield (Commercial) | Immediate chemical stabilization of nucleic acids upon sample contact, inhibiting nucleases and preventing degradation during mechanical processing. |
| CTAB Lysis Buffer | For plant/fungal tissues. Cetyltrimethylammonium bromide (CTAB) complexes with polysaccharides and contaminants during lysis, allowing their removal. |
| Proteinase K | A broad-spectrum serine protease used in conjunction with lysis buffers to digest proteins and nucleases, enhancing DNA release and stability. |
| Glycogen (Molecular Grade) | Acts as an inert carrier during ethanol/isopropanol precipitation, significantly improving the recovery and visibility of low-concentration DNA pellets. |
| SPRI (Solid Phase Reversible Immobilization) Beads | Magnetic beads that selectively bind DNA by size in PEG/NaCl buffer, enabling efficient purification, cleanup, and size selection post-lysis. |
| Phenol:Chloroform:Isoamyl Alcohol (25:24:1) | Organic extraction mixture that denatures and removes proteins, lipids, and other contaminants from the crude lysate. |
Technical Support Center: Troubleshooting Guides & FAQs
FAQ & Troubleshooting Section
Q1: During DNA extraction from a low-biomass soil sample, my final yield is consistently below the detection limit of a fluorometer. What are the primary causes and solutions? A: This is commonly due to DNA loss during extraction or inhibitor co-purification. Ensure you are using a kit specifically validated for low-biomass (e.g., Qiagen PowerSoil Pro Kit or MoBio DNeasy PowerLyzer). Incorporate a carrier RNA (like poly-A) during binding steps to minimize silica column loss. For inhibitor removal, a post-extraction clean-up with a kit like Zymo OneStep PCR Inhibitor Removal or a diluted CTAB wash can be effective. Always include an extraction blank to monitor contamination.
Q2: My extracts from formalin-fixed paraffin-embedded (FFPE) tissues show severe DNA fragmentation and I cannot amplify targets >200bp. How can I improve insert size? A: Formaldehyde causes cross-linking and fragmentation. Prior to standard extraction, de-crosslinking is essential. Protocol: Deparaffinize with xylene/ethanol washes. Incubate the sample in TE buffer (pH 9.0) with 1% SDS and 20mg/mL proteinase K at 65°C for 24-48 hours, refreshing reagents every 12 hours. Follow with a phenol-chloroform-isoamyl alcohol (25:24:1) extraction and use a repair enzyme mix (e.g., NEB PreCR Repair Mix or NEBNext FFPE DNA Repair Mix) for 1-2 hours at 37°C before purification.
Q3: I am getting high levels of modern human DNA contamination in my ancient bone powder extracts, swamping the endogenous signal. What steps can I take to mitigate this? A: Contamination is a critical issue. Implement strict physical isolation (dedicated clean room, UV hoods, positive pressure suits). Protocol: Surface-clean the bone with dilute bleach and UV irradiate all sides (254 nm, 15 min per side). Powder the inner compact bone. Use a digestion buffer with EDTA, SDS, and proteinase K for 24-48 hours at 37°C with constant rotation. Bind DNA to silica in the presence of guanidine thiocyanate and isopropanol. Consider USER enzyme treatment (for uracil-containing contaminant degradation) and use of bait-capture for enrichment.
Q4: My negative extraction controls for low-biomass sputum samples are showing positive amplification, indicating contamination. How do I diagnose the source? A: Systematically test each reagent and step. Create a matrix of control extractions: 1) Kit reagents only, 2) Sterile water processed through the full protocol, 3) Swabs of lab surfaces/equipment. Use a broad-range 16S rRNA PCR. If contamination persists, aliquot all liquids (buffers, water, ethanol) into single-use volumes, autoclave all non-enzymatic reagents, and use UV-treated plastics. Consider switching to a kit with minimal handling or one containing DNase to pre-treat reagents.
Q5: After repairing FFPE DNA, my sequencing library preparation still fails due to insufficient material. What quantification and library prep methods are most suitable? A: Fluorometers (Qubit) are more accurate than spectrophotometers (NanoDrop) for damaged DNA. Use a library prep kit designed for damaged/low-input DNA, such as the NEBNext Ultra II FS DNA Library Prep Kit or SMARTer ThruPLEX Plasma-Seq Kit. These incorporate steps to handle fragmented ends. A qPCR-based quantification method (like the KAPA Library Quantification Kit) for the final library is essential for accurate sequencing pool normalization.
Table 1: Comparison of DNA Yield from Different Difficult Sample Types Using Specialized Kits
| Sample Type | Typical Starting Material | Common Kit/Protocol | Average Yield (Range) | Key Inhibitors Removed |
|---|---|---|---|---|
| Low-Biomass (Swab) | 1 swab | PowerSoil Pro (Qiagen) | 0.05 - 0.5 ng/μL | Humic acids, polyphenols |
| FFPE Tissue (5μm slice) | 10 sections | GeneRead DNA FFPE (Qiagen) | 0.1 - 50 ng/μL (highly variable) | Formalin cross-links, proteins |
| Ancient Bone | 50 mg powder | Dabney et al. (2013) Silica-based | 0.001 - 0.1 ng/μL | Humics, collagen, salts |
| Formalin-Fixed Tissue (non-embedded) | 25 mg | Phenol-Chloroform + Repair | 1 - 100 ng/μL | Cross-links, proteins |
Table 2: Effect of Repair Enzymes on FFPE DNA Library Metrics
| Repair Treatment | Input DNA | % of Fragments >150bp | Library Conversion Efficiency | Post-Capture Duplication Rate |
|---|---|---|---|---|
| No Repair | 50 ng | 15% | 5-10% | >40% |
| UDG + Endo VIII (USER) | 50 ng | 22% | 10-15% | 30-40% |
| NEB PreCR Mix | 50 ng | 35% | 15-25% | 20-30% |
| NEBNext FFPE Repair | 50 ng | 45% | 25-35% | 15-25% |
Detailed Experimental Protocols
Protocol 1: Silica-Based DNA Extraction from Ancient Bone (Modified from Dabney et al., 2013)
- Surface Decontamination: Mechanically remove outer layer. Treat with 1-2% sodium hypochlorite (bleach) for 30 seconds, rinse with molecular-grade water. UV irradiate (254 nm, 15 min per side).
- Powdering: Using a sterile drill or mill, powder the inner compact bone in a dedicated clean room.
- Digestion: Incubate 50-100 mg powder in 1 mL digestion buffer (0.45M EDTA pH 8.0, 0.25 mg/mL Proteinase K, 0.05% SDS) at 37°C for 24-72h with rotation.
- Binding: Centrifuge. To supernatant, add 5x volume of PB buffer (Qiagen) or binding buffer (5M guanidine HCl, 40% isopropanol, 0.12M sodium acetate). Transfer to a silica spin column.
- Washing: Wash with PE buffer (Qiagen) or 80% ethanol.
- Elution: Elute in 50-100 μL of TE buffer (pH 8.0) or nuclease-free water.
Protocol 2: De-Crosslinking and Repair of FFPE-DNA
- Deparaffinization: Add 1 mL xylene to 10-20 μm sections, vortex, centrifuge. Discard supernatant. Repeat. Wash twice with 1 mL 100% ethanol. Air dry pellet.
- De-Crosslinking: Resuspend in 200 μL digestion buffer (100mM Tris-Cl pH 9.0, 1% SDS, 20mg/mL Proteinase K). Incubate at 65°C for 24h, adding fresh Proteinase K after 12h.
- Purification: Add 200 μL phenol:chloroform:isoamyl alcohol (25:24:1), vortex, centrifuge. Transfer aqueous phase to a new tube. Precipitate with 1/10 volume 3M sodium acetate and 2 volumes 100% ethanol. Wash with 70% ethanol.
- DNA Repair: Resuspend pellet in 50 μL TE. Add 10 μL NEBNext FFPE DNA Repair Buffer and 5 μL Repair Mix. Incubate at 20°C for 15 min, then 65°C for 15 min. Purify using AMPure XP beads.
Workflow & Pathway Visualizations
Title: FFPE DNA Extraction and Repair Workflow
Title: Low-Biomass Contamination Troubleshooting Path
The Scientist's Toolkit: Research Reagent Solutions
| Item | Primary Function | Application Note |
|---|---|---|
| Carrier RNA (e.g., Poly-A) | Binds to silica membrane, reducing loss of minute target DNA during wash steps. | Critical for low-biomass extractions; add to binding buffer. |
| Proteinase K (Molecular Grade) | Digests proteins and nucleases, critical for lysing cells and freeing DNA from complexes. | Essential for FFPE and ancient samples; use high concentrations and long incubations. |
| Guanidine Thiocyanate / HCl | Chaotropic salt that denatures proteins, inhibits nucleases, and promotes DNA binding to silica. | Core component of most modern silica-based extraction buffers. |
| Silica-Membrane Spin Columns | Selective binding of DNA in the presence of chaotropic salts, allowing purification from inhibitors. | Choose kits with minimal dead volume to maximize low-concentration elution. |
| AMPure XP / SPRI Beads | Magnetic beads that selectively bind DNA by size in PEG/NaCl buffer, enabling clean-up and size selection. | Used for post-extraction clean-up and NGS library preparation. |
| UDG / USER Enzyme Mix | Removes uracil bases (common in ancient and damaged DNA) and cleaves the abasic site, reducing contamination and damage. | Used in ancient DNA workflows to degrade modern contaminant DNA and remove damage. |
| NEBNext FFPE DNA Repair Mix | Enzyme cocktail (e.g., glycosylases, lysses, polymerases) that reverses common formalin-induced damage. | Specifically designed to repair FFPE-derived DNA fragments before library prep. |
| Humin/Inhibitor Removal Solution | Chemical agents (e.g., CTAB, PTB) that bind and precipitate organic inhibitors common in soil and plants. | Added during lysis step for challenging environmental samples. |
Troubleshooting Guides & FAQs
Q1: I am extracting DNA from a low-biomass soil metagenomic sample. My yields are consistently low and variable, leading to library prep failure for short-read sequencing. What should I do? A: Low and variable yield is common with inhibitor-rich samples. Key steps:
- Increase Sample Input: Use a higher volume/amount of starting material if possible.
- Incorporate Robust Lysis: Use a combination of mechanical (e.g., bead beating) and enzymatic lysis.
- Optimize Inhibitor Removal: Perform post-extraction purification with kits designed for environmental samples (e.g., with silica or magnetic bead clean-up in the presence of high concentrations of guanidine HCl). Consider a secondary clean-up step.
- Use Inhibition-Resistant Enzymes: For library prep, select polymerases and ligases specifically engineered for high inhibitor tolerance.
Q2: After extracting DNA from a gut microbiome sample and proceeding to long-read (ONT/PacBio) library prep, I get no sequencing output. The QC shows high-molecular-weight DNA but very low concentration. A: This suggests DNA damage or the presence of specific inhibitors (e.g., salts, organics) that interfere with library enzymes.
- Assess DNA Integrity: Run genomic DNA on a pulsed-field gel or FEMTO Pulse system to confirm size and detect fragmentation/ damage.
- Perform a Repair Step: Use a pre-library prep enzymatic repair mix (e.g., NEBNext FFPE Repair) to address nicks and damaged ends common in extracted metagenomic DNA.
- Desalt Thoroughly: Ensure the final elution of extracted DNA is in a low-EDTA TE buffer or nuclease-free water. Use a size-selective purification bead clean-up (e.g., 0.45X-0.8X SPRI beads) to remove small fragments and salts simultaneously.
Q3: My short-read library prep from environmental DNA results in extremely high adapter dimer peaks (~125bp) on the Bioanalyzer. How can I mitigate this? A: High adapter dimer indicates inefficient purification of fragmented/end-prepped DNA prior to adapter ligation.
- Optimize Bead Clean-up Ratios: After fragmentation & end-prep, use a double-sided SPRI bead clean-up. First, use a high ratio (e.g., 1.8X) to remove small fragments. After recovering the supernatant, add beads to a lower ratio (e.g., 0.8X) to bind the target-sized DNA. Elute carefully.
- Quantify Precisely: Accurately quantify the DNA after end-prep using a fluorescence-based assay (Qubit). Use the recommended input mass for adapter ligation to maintain optimal stoichiometry.
- Use Ligation-Free Kits: For future work, consider switching to a transposase-based (tagmentation) library prep kit, which integrates fragmentation and adapter addition in a single step, virtually eliminating adapter-dimer formation.
Q4: For hybrid short/long-read sequencing of the same metagenomic extract, how do I split my DNA to preserve high molecular weight (HMW) for long-read while having enough for short-read? A: This requires careful handling post-extraction.
- Primary QC: First, quantify total yield and assess integrity on an agarose gel or TapeStation.
- Aliquot for Long-Read: Before any vortexing or pipetting that could shear DNA, gently aliquot the required mass (e.g., 1-3 µg for ONT) for long-read sequencing using wide-bore tips. Do not perform bead clean-up unless necessary.
- Remainder for Short-Read: The remaining sample can be used for short-read library prep. If the concentration is low, concentrate it using a vacuum concentrator (not speed-vac) or ethanol precipitation. Standard bead clean-ups can be applied.
Q5: I see significant bias in my metagenomic sequencing data towards certain GC-content genomes. Could this be introduced during the integrated extraction-to-library prep workflow? A: Yes, both extraction and library prep can introduce GC bias.
| Workflow Stage | Potential Cause of GC Bias | Mitigation Strategy |
|---|---|---|
| Cell Lysis | Differential lysis efficiency of gram-positive vs. gram-negative bacteria. | Use harsher, standardized mechanical lysis (bead beating) for all samples. |
| DNA Fragmentation (Short-Read) | Sonication or enzymatic fragmentation can under-represent extreme GC genomes. | Calibrate fragmentation to target a larger size range; use validated enzymatic kits. |
| PCR Amplification | Polymerases can amplify mid-GC content templates more efficiently. | Use high-fidelity, bias-resistant polymerases (e.g., KAPA HiFi). Minimize PCR cycles; prefer PCR-free protocols. |
| Library Size Selection | Bead-based size selection can deplete fragments with atypical structures. | Use gel-based size selection or carefully calibrate SPRI bead ratios. |
Experimental Protocols
Protocol 1: Integrated HMW DNA Extraction and Clean-up for Long-Read Sequencing
Title: Extraction of Inhibitor-Free High Molecular Weight DNA from Fecal Samples for Nanopore Sequencing. Principle: This protocol combines chemical lysis, mechanical disruption, and magnetic bead-based purification to recover long DNA fragments while removing PCR inhibitors common in stool.
- Weigh 200 mg of frozen fecal material into a Lysing Matrix E tube.
- Add 1 mL of Lysis Buffer SLX-Mlus (containing guanidine thiocyanate and sarcosyl).
- Add 50 µL of Proteinase K (20 mg/mL). Vortex briefly.
- Incubate at 55°C for 30 minutes with gentle agitation.
- Securely mount tubes on a bead beater and homogenize at 6.0 m/s for 45 seconds. Place on ice for 2 minutes.
- Centrifuge at 13,000 x g for 5 minutes at 4°C. Transfer supernatant to a new 2 mL tube.
- Add 1 volume of Binding Buffer SPX (containing isopropanol and guanidine HCl) to the supernatant. Mix by inversion.
- Transfer 700 µL to a tube containing Magnetic Beads Sera-Mag SpeedBeads. Mix by pipetting and incubate at room temp for 5 min.
- Place on a magnetic rack. Discard supernatant.
- Wash beads twice with 500 µL of Freshly Prepared 80% Ethanol.
- Air-dry beads for 5-7 minutes. Elute DNA in 50-100 µL of Low-EDTA TE Buffer (pH 8.0) by incubating at 55°C for 5 min.
- Perform a second clean-up using 0.45X SPRI beads to remove short fragments and salts. Elute in 30 µL TE Buffer.
- Quantify using Qubit dsDNA HS Assay. Assess size distribution via FEMTO Pulse or Pulsed-Field Gel Electrophoresis.
Protocol 2: PCR-Free Short-Read Library Prep from Metagenomic DNA
Title: Construction of PCR-Free Illumina Libraries from Soil DNA Extracts. Principle: This protocol uses enzymatic fragmentation and adapter ligation to minimize bias, followed by size selection to generate libraries ready for Illumina sequencing.
- Dilute 100-500 ng of input DNA in 50 µL of Low TE.
- Add 50 µL of NEBNext Ultra II FS Buffer. Mix and briefly centrifuge.
- Incubate in a thermocycler: 37°C for 15 min (fragmentation), 65°C for 30 min (end-repair & dA-tailing). Hold at 4°C.
- Perform double-sided SPRI bead clean-up:
- Add 144 µL (1.8X) of SPRIselect Beads to the 100 µL reaction. Mix. Incubate 5 min.
- Place on magnet. Transfer 150 µL of supernatant to a new tube.
- Add 60 µL (0.8X of original volume) of SPRIselect Beads to the supernatant. Mix. Incubate 5 min.
- Place on magnet. Discard supernatant.
- Wash beads twice with 200 µL of 80% ethanol.
- Air-dry. Elute in 23 µL of 10 mM Tris-HCl (pH 8.0).
- To the eluate, add 25 µL of Blunt/TA Ligase Master Mix and 2 µL of Diluted NEBNext Adaptor. Incubate at 20°C for 15 min.
- Add 3 µL of USER Enzyme to the ligation mix. Incubate at 37°C for 15 min.
- Perform a final size selection with 0.85X SPRIselect Beads to remove adapter dimer and select for inserts >300 bp.
- Elute final library in 25 µL of 10 mM Tris-HCl. Quantify by qPCR (KAPA Library Quant Kit). Pool and sequence.
Mandatory Visualizations
Title: Integrated Workflow for Short & Long-Read Sequencing from a Single Sample
Title: Troubleshooting Flow for Extraction & Library Prep Failures
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Workflow | Key Consideration for Metagenomics |
|---|---|---|
| Lysing Matrix E Tubes | Contains ceramic/silica beads for mechanical cell disruption. Essential for breaking gram-positive bacteria and fungal spores in environmental samples. | Ensure bead beating time is standardized across samples to avoid bias from differential lysis. |
| Magnetic Beads (SPRIselect/Sera-Mag) | Carboxyl-coated magnetic particles for size-selective binding and purification of DNA. Used in clean-up and size selection. | The bead-to-sample ratio is critical for size cut-off. Calibrate ratios for desired fragment retention (e.g., 0.45X for HMW, 0.8X for short-insert libraries). |
| Guanidine-Based Buffers | Chaotropic salts that denature proteins, inhibit nucleases, and promote nucleic acid binding to silica/magnetic beads. | High concentrations are needed for inhibitor-rich samples but must be thoroughly removed in wash steps to avoid interference with downstream enzymes. |
| NEBNext Ultra II FS Enzyme Mix | A two-enzyme mix for simultaneous fragmentation and end-repair/dA-tailing of DNA for Illumina library prep. | Reduces bias compared to sonication. Input DNA quality (lack of nicks) significantly impacts final fragment size distribution. |
| KAPA HiFi HotStart ReadyMix | A high-fidelity, bias-resistant PCR polymerase master mix. Used for amplifying low-input libraries. | Essential for minimizing GC bias during the optional PCR amplification step. Keep cycles to a minimum (≤8). |
| Low-EDTA TE Buffer (10:0.1) | Elution and storage buffer (10 mM Tris-HCl, 0.1 mM EDTA, pH 8.0). | The low EDTA concentration is crucial for long-read sequencing (e.g., Nanopore), as Mg²⁺ is a cofactor for sequencing enzymes. |
| DNA Repair Mix (e.g., NEB FFPE) | Enzyme mix to repair nicked, damaged, or fragmented DNA. Contains DNA polymerase, ligase, and kinase. | Critical pre-step for long-read library prep from environmental DNA, which often contains damage from extraction or sample storage. |
Technical Support Center
Troubleshooting Guides & FAQs
Q1: During automated DNA extraction, my 96-well plate yields consistently low DNA concentrations in columns 11 & 12. What could be the cause? A: This is a classic symptom of reagent depletion in automated liquid handlers. Verify that the reservoir volumes for lysis buffer and binding buffer are sufficient for the full plate run. For a standard silica-membrane plate protocol, ensure at least 1.2 mL of each reagent per column is available. Recalibrate the pipetting head for volume accuracy in those well positions. Check for clogged tips or blocked fluidics in the channels servicing those columns.
Q2: My extracted metagenomic DNA shows PCR inhibition in downstream assays, despite high 260/280 ratios. How can I troubleshoot this from the extraction stage? A: Inhibition often stems from carryover of humic acids (common in soil/stool samples) or chaotropic salts. First, review your wash steps: ensure you are using the recommended volumes of wash buffer (typically Buffer AW1 and AW2 for QIAamp 96 kits) and that the centrifugation steps are at correct speed/time (e.g., 5600 x g, 1 min). Consider integrating an additional inhibitor removal wash step (see protocol below). Quantify inhibition using a spike-in qPCR control.
Q3: When scaling from manual extraction to an automated platform (e.g., QIAcube HT, KingFisher), my DNA fragment size distribution is shorter. How do I preserve high molecular weight DNA? A: Automated systems can introduce more shear stress. To mitigate:
- Lysis Optimization: Ensure bead-beating steps (if used) are performed in a horizontal, orbital motion at a fixed, optimized speed (e.g., 1800 rpm) and duration (e.g., 45s), not vortexing.
- Liquid Handling: Adjust pipetting parameters to reduce speed and avoid air bubble introduction during mixing and transfer.
- Magnetic Bead Handling: For magnetic bead-based protocols, reduce incubation time with beads post-binding to minimize co-purification of sheared fragments.
Detailed Experimental Protocols
Protocol: Automated High-Throughput DNA Extraction from Fecal Samples using Magnetic Bead Technology (KingFisher System)
Objective: To reproducibly extract inhibitor-free microbial DNA from 96 fecal samples for shotgun metagenomic sequencing.
Materials: Pre-filled deep-well plate with 750 µL of lysis buffer (500mM NaCl, 50mM Tris-HCl pH 8.0, 50mM EDTA, 4% SDS), proteinase K, Sera-Mag SpeedBeads (carboxylated magnetic beads), 80% Ethanol, TE buffer.
Methodology:
- Homogenization & Lysis: Aliquot 100mg of fecal sample into lysis plate. Add 20 µL proteinase K. Seal and incubate at 56°C for 1 hour with shaking at 1000 rpm.
- Binding: Add 40 µL of magnetic bead suspension to each well. Mix thoroughly by pipetting. Incubate at room temperature for 10 minutes to allow DNA binding.
- Automated Purification (KingFisher Program):
- Wash 1: Transfer beads to 1 mL of wash buffer 1 (Guanidine HCl + Isopropanol). Mix for 2 minutes.
- Wash 2: Transfer beads to 1 mL of wash buffer 2 (80% Ethanol). Mix for 1 minute. Repeat once.
- Elution: Transfer beads to 100 µL of pre-heated (65°C) TE Buffer (10 mM Tris-HCl, 1mM EDTA, pH 8.0). Incubate at 65°C for 5 minutes with mixing to elute DNA.
- Bead Separation & Storage: The KingFisher magnet collects beads, and the eluate is transferred to a fresh plate. Quantify DNA using a fluorescence assay (e.g., Qubit dsDNA HS Assay). Store at -80°C.
Protocol: Integration of an Additional Inhibitor Removal Step
After the initial lysis and before magnetic bead binding, add 250 µL of inhibitor removal solution (e.g., 1M Potassium Phosphate buffer, pH 8.0). Vortex for 30s, incubate on ice for 5 min, then centrifuge at 13,000 x g for 5 min. Transfer the supernatant to a new well for the binding step. This precipitates many humic acids.
Data Presentation
Table 1: Comparison of Automated DNA Extraction Platforms for Metagenomic Studies
| Platform (Example) | Principle | Avg. Yield (Stool, ng/mg) | Avg. 260/280 | Avg. Fragment Size (bp) | Hands-On Time (for 96 samples) | Inhibition Rate (Failed qPCR) |
|---|---|---|---|---|---|---|
| KingFisher Apex | Magnetic Beads | 45 ± 12 | 1.85 ± 0.1 | >10,000 | ~1.5 hours | <5% |
| QIAcube HT | Silica-Membrane Plate | 38 ± 15 | 1.90 ± 0.05 | 5,000 - 8,000 | ~1 hour | 8-12% |
| MagMAX Core HT | Magnetic Beads | 42 ± 10 | 1.87 ± 0.08 | >9,000 | ~2 hours | <3% |
Table 2: Troubleshooting Common Yield and Quality Issues
| Symptom | Possible Cause | Corrective Action |
|---|---|---|
| Low yield across entire plate | Inefficient cell lysis | Increase lysis incubation time; optimize bead-beating parameters; add mechanical lysis step. |
| High 260/230 ratio (<1.7) | Carryover of organic compounds (phenol, guanidine) | Ensure complete removal of Wash Buffer 1; add an extra 80% ethanol wash step. |
| High variability between replicates | Inconsistent sample input or reagent mixing | Standardize sample homogenization; increase mixing cycles/ speed during binding and wash steps. |
| PCR inhibition despite good yield | Humic acid or salt carryover | Integrate the inhibitor removal protocol step; dilute DNA template 1:10 in downstream assay. |
Visualizations
Title: Automated DNA Extraction Workflow for Metagenomics
Title: Troubleshooting Low Yield in Edge Wells
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for High-Throughput Metagenomic DNA Extraction
| Item | Function | Example/Composition Key Note |
|---|---|---|
| Lysis Buffer (with SDS & EDTA) | Disrupts cell membranes & inactivates nucleases. High pH aids dissociation from proteins. | Contains 4% SDS, 50mM EDTA, pH ~8.0. Crucial for Gram-positive bacteria. |
| Proteinase K | Proteolytic enzyme that digests proteins and aids in complete lysis. | Must be quality-controlled for RNase/DNase-free activity. Added at >50 mAU/mL. |
| Carboxylated Magnetic Beads | Bind DNA via salt-bridging in high chaotropic salt conditions. Enable automation. | Sera-Mag SpeedBeads. Size uniformity is critical for reproducible binding capacity. |
| Chaotropic Salt Solution (Binding Buffer) | Disrupts hydrogen bonding; makes DNA hydrophobic so it binds to silica/beads. | Typically 4-6M Guanidine HCl. Concentration directly impacts yield. |
| Ethanol-Based Wash Buffer | Removes salts, proteins, and other contaminants while keeping DNA bound. | 80% Ethanol is standard. Must be freshly prepared to avoid hydration. |
| Inhibitor Removal Solution | Precipitates or sequesters specific inhibitors like humic acids. | 1M Potassium Phosphate, pH 8.0, or commercial kits like Zymo Research's Inhibitor Removal Technology. |
| Low-EDTA TE Elution Buffer | Chelates Mg2+ to inhibit nucleases but low EDTA avoids sequencer interference. | 10 mM Tris-HCl, 0.1 mM EDTA, pH 8.0-8.5. Pre-heating to 65°C increases yield. |
| RNase A (Optional) | Degrades RNA to prevent overestimation of DNA yield and clogging of spin columns. | Used if extracting total nucleic acid. Must be heat-inactivated if needed for RNA work. |
Solving Common Pitfalls: Troubleshooting and Optimizing Your DNA Yield and Purity
Troubleshooting Guide
Q1: Why is my DNA yield from a metagenomic soil sample so low? A: Low yield is often due to inefficient cell lysis or inhibitor co-purification. For complex samples like soil, mechanical lysis (e.g., bead beating) is crucial but must be optimized. Inadequate inhibitor removal (humic acids, polyphenols) can also reduce measurable yield by interfering with downstream quantification.
Q2: What causes excessive DNA shearing during extraction, and why is it a problem for metagenomics? A: Excessive shearing results from overly vigorous mechanical lysis (e.g., excessive bead beating time/speed) or harsh chemical/ enzymatic lysis. For metagenomic research, shearing fragments below 10 kb can compromise the assembly of long genomic contigs, hinder binning of complex communities, and limit the detection of large gene clusters.
FAQs
Q3: How can I quickly diagnose the primary cause of low yield? A: Perform a stepwise diagnostic protocol:
- Post-Lysis Visualization: Check a small aliquot of lysate under a phase-contrast microscope to confirm cell disruption.
- Inhibitor Test: Perform a spiked PCR with a known quantity of control DNA alongside your extracted sample. Inhibition is indicated if the spiked control amplifies poorly in the sample extract but well in buffer.
- Carrier RNA Test: Re-extract using a carrier RNA (like poly-A RNA). A significant yield increase suggests losses were due to low-abundance DNA binding inefficiently to silica columns.
Q4: What are the best corrective actions if I suspect both low yield and shearing? A: Optimize the lysis step. A balanced protocol is required. The table below summarizes key parameters:
Table 1: Optimization of Bead-Beating for Metagenomic DNA Extraction
| Parameter | Default (Typical Issue) | Optimized for Yield & Size | Rationale |
|---|---|---|---|
| Bead Size | 0.1 mm glass beads (High shearing) | Mix of 0.5 mm and 0.1 mm beads | Larger beads for cell clustering disruption, smaller for efficient lysis. |
| Bead Beating Time | 10 min (High shearing) | 2-4 minutes (titer required) | Minimizes physical shearing while maintaining lysis efficiency. |
| Lysis Buffer | Standard SDS-based | Buffer with added CTAB & Proteinase K | CTAB complexes with polysaccharides and humics; Proteinase K digests proteins. |
| Post-Lysis Incubation | Immediate processing | 10 min at 55°C after beating | Allows chemical/enzymatic lysis to complete, reducing needed physical force. |
| Post-Lysis Clarification | None (Inhibitor carryover) | Centrifugation + supernatant filtration (5 µm) | Removes debris and residual beads that can shear DNA during pipetting. |
Experimental Protocol: Inhibitor Removal with CTAB and Size Selection
Title: Combined CTAB and Gel-Based Size Selection for High-Molecular-Weight Metagenomic DNA.
- Lysis: Process sample (0.5 g soil) with optimized bead-beating (Table 1) in Tris-EDTA-SDS buffer with 1% CTAB.
- Purification: Perform phenol:chloroform:isoamyl alcohol (25:24:1) extraction. Centrifuge at 12,000 x g for 10 min at 4°C. Transfer aqueous phase.
- Precipitation: Add 0.7 volumes isopropanol and 0.1 volumes 3M sodium acetate (pH 5.2). Precipitate at -20°C for 1 hour. Pellet DNA at 12,000 x g for 30 min.
- Wash & Resuspend: Wash pellet with 70% ethanol. Air-dry and resuspend in TE buffer (pH 8.0).
- Size Selection: Load DNA onto a low-melting-point agarose gel (0.5-0.7%). Excise the high-molecular-weight region (>10 kb) using a clean scalpel.
- DNA Recovery: Purify DNA from the gel slice using GELase or β-agarase enzyme according to manufacturer protocol.
Visualizing the Diagnostic Workflow
Title: Diagnostic and Corrective Action Flow for DNA Yield & Shearing
The Scientist's Toolkit: Key Reagent Solutions
Table 2: Essential Reagents for HMW Metagenomic DNA Extraction
| Item | Primary Function in Context | Key Consideration |
|---|---|---|
| Inhibitor Removal Technology (IRT) Beads / Columns | Bind to humic acids, polyphenols, and other common environmental inhibitors during purification. | Essential for soil and sediment samples. Often integrated into commercial kits. |
| Guanidine Hydrochloride (GuHCl) | A chaotropic salt that denatures proteins, aids in cell lysis, and promotes DNA binding to silica. | Preferred over guanidine thiocyanate for very long DNA as it is less degrading. |
| Carrier RNA (e.g., Poly-A) | Co-precipitates with and "carries" trace amounts of DNA, reducing losses during silica column binding or ethanol precipitation. | Critical for low-biomass samples. Must be RNase-free. |
| Proteinase K | Broad-spectrum serine protease that digests nucleases and other proteins, aiding lysis and protecting DNA. | Incubation at 55°C after mechanical lysis is highly effective. |
| CTAB (Cetyltrimethylammonium Bromide) | Precipitates polysaccharides and complexes with humic acids, allowing their removal during extraction. | Used in pre-lysis buffers for plant-rich or humic-rich soils. |
| β-Agarase / GELase | Enzymes that digest agarose, allowing recovery of DNA from low-melt agarose gels after size selection. | Enables purification of HMW DNA away from sheared fragments and residual inhibitors. |
| Magnetic Beads (SPRI) | Polyethylene glycol (PEG)-coated beads for selective binding and size selection of DNA fragments. | Bead-to-sample ratio can be adjusted for crude size selection (e.g., >1kb). |
| TE Buffer (pH 8.0) | Resuspension buffer. Tris maintains pH, EDTA chelates Mg2+ to inhibit DNases. | pH 8.0 is crucial for long-term DNA stability and accurate quantitation. |
Technical Support Center: Troubleshooting & FAQs
Q1: My post-column eluted DNA has low yield but high purity (A260/A280 ~1.8). Was inhibitor removal too aggressive? A: Likely yes. Over-efficient polyphenol/complex polysaccharide binding can co-precipitate or adsorb DNA. Optimize:
- PVPP: Reduce amount from standard 2% (w/v) to 0.5-1% in initial lysis buffer. Pre-wash PVPP with buffer to remove fine particles.
- CTAB: Ensure extraction is performed above the critical micelle temperature (≥60°C). For tough samples, use a sequential CTAB protocol (Table 1).
- Protocol: Sequential CTAB-PVPP Lysis for Complex Samples:
- Homogenize sample in CTAB Lysis Buffer (2% CTAB, 1.4M NaCl, 100mM Tris-HCl pH 8.0, 20mM EDTA) at 65°C for 30 min.
- Add an equal volume of PVPP Solution (1% w/v in sterile water, pre-washed) to the supernatant post initial centrifugation (12,000xg, 10 min).
- Incubate on ice for 15 min, then centrifuge (12,000xg, 15 min).
- Proceed with chloroform:isoamyl alcohol (24:1) extraction on the supernatant.
Q2: I see a viscous, gelatinous pellet after CTAB precipitation. How do I recover DNA and proceed? A: The gelatinous pellet contains CTAB-polysaccharide complexes entangled with DNA. Do NOT discard.
- Dissolve the pellet in High-Salt TE Buffer (10mM Tris-HCl, 1mM EDTA, 1M NaCl) by gentle pipetting and incubation at 65°C for 15-30 min.
- Add 0.6 volumes of isopropanol to precipitate DNA selectively, leaving polysaccharides in solution. Incubate at -20°C for 1 hour.
- Centrifuge at 12,000xg, 4°C, for 20 min. Wash pellet with 70% ethanol (in high-salt: 0.2M NaCl) and resuspend in low-salt TE buffer.
Q3: My column-based purification post-CTAB/PVPP yields DNA that inhibits downstream PCR. What wash buffer optimizations are critical? A: Residual CTAB or humic acids are likely carried over. Standard silica-column wash buffers (e.g., 80% ethanol) may be insufficient. Implement an optimized wash regime (Table 2).
- Protocol: Optimized Three-Step Column Wash:
- Wash 1 (Desalting): Apply 800µL of Wash Buffer A (5mM Tris-HCl pH 7.5, 80% Ethanol, 20mM NaCl). Centrifuge at full speed for 30 sec. Discard flow-through.
- Wash 2 (Inhibitor Removal): Apply 700µL of Wash Buffer B (100mM Sodium Acetate pH 5.0, 80% Ethanol). Let it sit on the column for 2 min, then centrifuge. This acidic wash helps dissociate humic acids.
- Wash 3 (Drying): Perform a second centrifugation with an empty collection tube for 2 min to dry the membrane completely before elution.
Q4: How do I choose between adding PVPP to the lysis buffer vs. post-lysis supernatant? A: The choice depends on the sample's primary inhibitor (Table 3).
Table 1: Sequential vs. Single CTAB-PVPP Protocol Yield/Purity Comparison
| Protocol | Sample Type | Avg. DNA Yield (ng/g) | A260/A280 | A260/A230 | PCR Success Rate |
|---|---|---|---|---|---|
| Single CTAB (2%) | Peat Soil | 45 ± 12 | 1.65 ± 0.08 | 1.2 ± 0.3 | 40% |
| Sequential CTAB-PVPP | Peat Soil | 68 ± 15 | 1.78 ± 0.05 | 1.8 ± 0.2 | 95% |
| Single CTAB (2%) | Plant Rhizosphere | 210 ± 45 | 1.72 ± 0.06 | 1.5 ± 0.4 | 75% |
| CTAB + In-Buffer PVPP (1%) | Plant Rhizosphere | 180 ± 30 | 1.81 ± 0.04 | 1.9 ± 0.1 | 98% |
Table 2: Impact of Optimized Column Wash on Inhibitor Removal
| Wash Regime | Residual CTAB (ng/µL)* | Humic Acid (Abs 340nm)* | Library Prep Success (NGS) |
|---|---|---|---|
| Standard (80% Ethanol, 2x) | 15.2 ± 3.1 | 0.25 ± 0.05 | 3/10 |
| Optimized 3-Step | 2.1 ± 0.8 | 0.05 ± 0.01 | 9/10 |
| *Measured in final 50µL eluate from a spiked control purification. |
Table 3: PVPP Addition Strategy Guide
| Addition Point | Target Inhibitor Class | Mechanism | Best For Sample Types |
|---|---|---|---|
| In Lysis Buffer | Polyphenols, Tannins | Binds inhibitors as they are released, preventing oxidation and complexation with DNA. | Fresh plant tissue, medicinal herbs, compost. |
| Post-Lysis to Supernatant | Humic Acids, Fulvic Acids | Binds soluble humics after cell debris removal, reducing competition for silica column binding. | Mature soils (peat, forest), sediment, sludge. |
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Inhibitor Removal |
|---|---|
| PVPP (Cross-linked) | Insoluble polymer that binds polyphenols/tannins via hydrogen bonds, preventing quinone formation and DNA coprecipitation. |
| CTAB (Cetyltrimethylammonium bromide) | Cationic detergent that complexes anionic polysaccharides (e.g., pectin, xylan) and acidic polyphenols in high-salt conditions, precipitating them. |
| High-Salt TE Buffer (1M NaCl) | Dissolves CTAB-polysaccharide pellets and prevents premature DNA binding to silica, allowing selective precipitation. |
| Sodium Acetate Wash Buffer (pH 5.0) | Acidic, salt-containing ethanol wash promotes dissociation and removal of humic acids from silica membrane. |
| Silica Membrane Columns | Selective binding of DNA in high-salt, elution in low-salt. Optimized washes are key for final purity. |
| Chloroform:Isoamyl Alcohol (24:1) | Denatures and removes proteins, lipids, and residual CTAB micelles via phase separation. |
Experimental Workflow & Decision Pathway
Title: DNA Extraction & Inhibitor Removal Decision Pathway
Title: Mechanism of Optimized Acidic Column Wash
Troubleshooting Guides & FAQs
Q1: My post-extraction DNA yield is low, but bead-beating was vigorous. What could be wrong? A: High-intensity mechanical lysis can fragment DNA excessively, leading to loss during silica-column binding (fragments <300 bp bind inefficiently). Verify fragment size distribution on a Bioanalyzer. If the median size is below 500 bp, reduce bead-beating time or intensity. For example, shift from a 5-minute continuous beat to 3 cycles of 1 minute beating with 1-minute rests on ice.
Q2: I need high-molecular-weight (>20 kb) DNA for long-read sequencing, but my soil sample lysis is inefficient. How can I improve yield without shearing? A: Prioritize enzymatic and chemical lysis over mechanical. Use an extended, stepped protocol: 1) Pre-treatment with Chelex-100 and EDTA to chelate nucleases. 2) Incubate with lysozyme (2 hr, 37°C), then proteinase K with SDS (2 hr, 56°C). 3) A gentle manual inversion mix with 0.1mm beads (no vortex). Avoid phenol-chloroform if possible; use high-salt precipitation.
Q3: My fragment length distribution is bimodal after lysis. What does this indicate? A: A bimodal distribution often indicates incomplete lysis of a subset of cells (e.g., Gram-positive bacteria or spores) alongside complete lysis of others. The larger mode is from the resistant cells that finally lysed under harsh conditions, fragmenting heavily. Implement a differential lysis protocol: gentle enzymatic lysis first to recover DNA from easy-to-lyse cells (separate supernatant), followed by mechanical lysis for the pellet.
Q4: How do I objectively balance lysis settings for a novel sample type (e.g., biofilm)? A: Perform a controlled Lysis Matrix Experiment. Vary one key parameter at a time (e.g., bead-beating time) and measure two outputs: Total DNA Yield (Qubit) and Average Fragment Length (TapeStation). The optimal balance is the point where further increases in lysis intensity produce negligible yield gains but cause significant fragment length reduction.
Data from a Representative Lysis Matrix Experiment (Soil Sample):
| Bead-Beating Time (min) | Total DNA Yield (ng/µl) | Average Fragment Length (bp) | Metagenomic Coverage (%)
| 0 (Enzymatic only) | 15.2 ± 2.1 | 12,500 ± 1,800 | 45 ± 5
| 1 | 42.5 ± 3.8 | 8,200 ± 950 | 78 ± 4
| 3 | 55.1 ± 4.2 | 3,500 ± 420 | 95 ± 3
| 5 | 56.3 ± 5.0 | 1,200 ± 150 | 92 ± 6
| 10 | 52.8 ± 6.1 | 450 ± 80 | 85 ± 7
Protocol for the Lysis Matrix Experiment:
- Sample Aliquot: Divide homogenized sample into 5x 0.5g aliquots.
- Lysis Variation: Use a standard buffer (e.g., Tris-EDTA-SDS). Process each aliquot in a bead-beater with 0.1mm silica/zirconia beads for the target time (0, 1, 3, 5, 10 min). Keep temperature at 4°C.
- Post-Lysis: Centrifuge. Split supernatant: one part for yield/size analysis, one part for standardized purification (same SPRI bead ratio for all).
- Quantification & QC: Measure yield (fluorometry) and size profile (automated electrophoresis). Perform qPCR on a conserved 16S rRNA gene region to estimate microbial coverage.
Q5: My downstream library preparation for shotgun sequencing fails after efficient lysis. Why? A: Excessive lysis can release high concentrations of humic acids, polysaccharides, or proteins that co-precipitate with DNA. These inhibitors can block library assembly enzymes. If you have high yield but poor library conversion, assess purity (A260/A230 ratio). Values below 2.0 indicate contamination. Implement a cleanup step with inhibitor-removal columns or CTAB-based purification before library prep.
Visualizing the Decision Framework
Title: Decision Workflow for Lysis Strategy Selection
Title: Relationship Between Lysis Intensity and Key Outcomes
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Primary Function in Balancing Lysis |
|---|---|
| Zirconia/Silica Beads (0.1mm, 0.5mm) | Mechanical shearing agent. Smaller beads (0.1mm) provide more efficient but harsher lysis. A mix can improve efficiency for diverse cell walls. |
| Lysis Buffer with EDTA & SDS | Chemical lysis. EDTA chelates Mg2+, inhibiting DNases. SDS dissolves lipid membranes and denatures proteins. |
| Proteinase K | Serine protease. Degrades cellular proteins and nucleases, crucial for gentle enzymatic lysis steps. |
| Lysozyme | Enzyme that hydrolyzes peptidoglycan in bacterial cell walls, especially effective for Gram-positive cells. |
| SPRI (Solid Phase Reversible Immobilization) Beads | Magnetic beads that bind DNA based on size. Critical for post-lysis size selection to remove very short fragments. |
| Inhibitor Removal Technology Columns (e.g., PVPP, CTAB) | Removes humic acids, polyphenols, and polysaccharides that are co-released during harsh lysis and inhibit enzymes. |
| Guanidine Hydrochloride/ Thiocyanate | Chaotropic salt. Disrupts cells, denatures proteins, and, at high concentrations, aids in nucleic acid binding to silica. |
| Phenol-Chloroform-Isoamyl Alcohol | Organic extraction. Removes proteins and lipids. Effective but can shear DNA; use with caution for HMW goals. |
Technical Support & Troubleshooting Center
This support center addresses common issues encountered during QC of metagenomic DNA extraction using fluorometry, gel electrophoresis, and qPCR.
Fluorometry Troubleshooting Guide
Q1: My fluorometry readings (Qubit/Broad Range assay) show high DNA concentration, but the sample fails downstream PCR. What could be wrong? A: This discrepancy often indicates the presence of inhibitory substances co-extracted with DNA (e.g., humic acids, phenolic compounds, salts) that interfere with the fluorescent dye binding. The fluorometer measures only double-stranded DNA that successfully binds the dye, but inhibitors may not affect this initial binding. Verify by diluting the sample (1:5, 1:10) and re-measuring; if the concentration does not drop proportionally, inhibitors are likely present. Perform a purification clean-up step (e.g., using silica-column kits designed for inhibitor removal) and re-quantify.
Q2: The fluorometer gives inconsistent readings between replicates of the same sample. A: This is typically a pipetting error or incomplete mixing. Ensure the sample and the fluorometric dye reagent are thoroughly mixed by vortexing for 3-5 seconds after combination. Use calibrated pipettes and low-binding tips for viscous metagenomic samples. Always prepare a fresh standard curve for each run, and ensure the assay tube is free of bubbles before reading.
Gel Electrophoresis Troubleshooting Guide
Q3: My post-extraction gel shows a smear with no distinct high-molecular-weight band. A: A smear indicates significant DNA shearing or degradation. Pre-extraction causes include excessive bead-beating duration or speed during cell lysis for tough environmental samples. Post-extraction causes are overly vigorous pipetting or multiple freeze-thaw cycles. Optimize lysis conditions (e.g., reduce bead-beating time in steps of 30 seconds) and always handle DNA with wide-bore tips. Include a positive control of intact genomic DNA (e.g., lambda phage DNA) on the gel.
Q4: The gel shows a bright, fast-migrating band, but the fluorometer shows low yield. A: This suggests a high concentration of RNA contamination. RNA stains intensely with intercalating dyes (e.g., Ethidium Bromide, SYBR Safe) and migrates quickly, creating a bright band near the dye front. Treat the extract with RNase A (heat-labile to allow later RNA-seq if needed), re-purify, and re-analyze.
qPCR Troubleshooting Guide
Q5: My qPCR amplification curves for the bacterial 16S rRNA gene are erratic or show very late Ct values in extracted samples, despite good fluorometry readings. A: This confirms the presence of PCR inhibitors. Run a dilution series of your sample. If the Ct values improve with dilution, inhibitors are present. Incorporate a internal control (exogenous DNA spiked into the sample pre-extraction) to distinguish between inhibition and low target abundance. Use inhibitor-tolerant polymerases or perform additional clean-up.
Q6: The no-template control (NTC) in my qPCR assay shows amplification. A: This indicates contamination, most likely from amplicon carryover or contaminated reagents. Use separate, dedicated pre- and post-PCR workspaces and pipettes. Prepare all master mixes in a UV-treated laminar flow hood. Aliquot reagents to avoid repeated freeze-thaw cycles. Use uracil-DNA glycosylase (UDG) and dUTP in your master mix to prevent re-amplification of carryover products.
Frequently Asked Questions (FAQs)
Q: What is the most critical QC checkpoint for metagenomic sequencing library prep? A: The post-extraction qPCR for a universal marker gene (e.g., 16S rRNA for bacteria) is the most critical functional assay. It confirms that the DNA is not only present and intact (per fluorometry and gel) but also amplifiable and free of inhibitors that would undermine library preparation and sequencing.
Q: Can I skip gel electrophoresis if I have fluorometry and qPCR data? A: It is not recommended. Gel electrophoresis provides visual confirmation of DNA integrity (high molecular weight) and alerts you to issues like RNA contamination or severe degradation that might not be apparent from the other two methods. It is a quick, low-cost checkpoint.
Q: How do I choose between a broad-range and high-sensitivity fluorometry assay? A: Use the broad-range assay (e.g., Qubit BR) for initial post-extraction quantification of expected high-yield samples. Use the high-sensitivity assay (e.g., Qubit HS) for low-biomass samples or for quantifying diluted DNA pre-library preparation.
Q: What should I use as a positive control for qPCR in diverse metagenomic samples? A: Spike a known, low-concentration of control DNA (from an organism not expected in your sample, e.g., Arabidopsis thaliana) into your lysis buffer before extraction. Its recovery and Ct value post-extraction directly measure extraction efficiency and inhibitor presence.
Table 1: Expected QC Metrics for High-Quality Metagenomic DNA
| QC Method | Optimal Result (Pre-Library Prep) | Acceptable Range | Action Required If Outside Range |
|---|---|---|---|
| Fluorometry (dsDNA HS) | Yield > 1 ng/µL | 0.5 - 100 ng/µL | Concentrate if too low; Dilute/clean if too high. |
| 260/280 (Nanodrop) | ~1.8 | 1.7 - 2.0 | If <1.7, protein/phenol contamination. Purify. |
| 260/230 (Nanodrop) | ~2.0 | 1.8 - 2.2 | If <1.8, humic acid/salt contamination. Purify. |
| Gel Electrophoresis | Single, tight HMW band (>10 kb) | Visible HMW smear | If intense low-MW smear, RNase treat. If degraded, re-extract. |
| qPCR (16S Ct) | Ct < 30 (for 1 ng template) | Ct < 35 | If Ct > 35 or amplification failure, suspect inhibition. Dilute/clean. |
Table 2: Troubleshooting Matrix for Common QC Failures
| Symptom | Fluorometry | Gel | qPCR | Most Likely Cause | Corrective Action |
|---|---|---|---|---|---|
| Low yield | Low conc. | Faint/No band | High Ct/No amp. | Inefficient cell lysis or DNA binding. | Increase lysis rigor (e.g., enzyme cocktail, longer bead-beating). |
| Inhibition | Normal/High | Normal HMW band | High Ct/No amp. | Co-purified inhibitors. | Dilute template 1:10. Use inhibitor-removal kit. |
| Degradation | Low/Normal | LMW smear | High Ct (if amp.) | Shearing during extraction/handling. | Gentler pipetting (wide-bore tips). Avoid freeze-thaw. |
| RNA Contam. | Normal/Low | Bright low-MW band | Normal | RNase not used/ineffective. | Treat with RNase A, re-purify. |
| Protein Contam. | Normal | Normal | Normal | Incomplete proteinase K digestion. | Add fresh proteinase K, extend digestion time. |
Detailed Experimental Protocols
Protocol 1: Integrated QC Workflow for Metagenomic DNA
Purpose: To assess the quantity, quality, purity, and functionality of DNA extracted from a complex environmental sample (e.g., soil, water filtrate).
Post-Extraction Fluorometry:
- Use the Qubit dsDNA High Sensitivity (HS) Assay kit.
- Prepare working solution by diluting Qubit reagent 1:200 in buffer.
- Prepare standards #1 and #2 as per kit.
- For samples: Mix 1 µL of extracted DNA with 199 µL of working solution (1:200 dilution) in a Qubit assay tube.
- Vortex, incubate 2 minutes at room temperature, protected from light.
- Read on Qubit fluorometer using the "dsDNA HS" setting.
- Calculation: The instrument outputs concentration (ng/µL). Multiply by elution volume and dilution factor for total yield.
Post-Extraction Gel Electrophoresis:
- Prepare a 0.8% agarose gel in 1X TAE buffer with a safe DNA stain (e.g., SYBR Safe, 1X final concentration).
- Mix 5 µL of DNA sample with 1 µL of 6X loading dye.
- Load alongside a DNA ladder suitable for high molecular weight (e.g., Lambda HindIII).
- Run gel at 4-5 V/cm for 45-60 minutes in 1X TAE buffer.
- Visualize using a blue-light gel imager. Assess for a single, high-molecular-weight band (>10 kb) and absence of a bright, fast-migrating RNA band.
Post-Extraction qPCR for Amplifiability:
- Target: Universal bacterial 16S rRNA gene (e.g., primers 341F/518R).
- Use an inhibitor-resistant master mix (e.g., with BSA).
- Reaction Setup (20 µL):
- 10 µL 2X Master Mix
- 0.8 µL Forward Primer (10 µM)
- 0.8 µL Reverse Primer (10 µM)
- 0.4 µL 50X ROX Reference Dye (if required)
- 2 µL DNA Template (diluted to ~1 ng/µL based on Qubit)
- Nuclease-free water to 20 µL
- Include a standard curve (e.g., 10^1 to 10^6 copies of a cloned 16S fragment) and a no-template control (NTC).
- Cycling Conditions: 95°C for 3 min; 40 cycles of 95°C for 15 sec, 55°C for 30 sec, 72°C for 30 sec (with plate read); Melt curve analysis.
- Analysis: Determine Ct values. The sample should amplify within the linear range of the standard curve. A Ct > 35 for 1 ng of template suggests inhibition or low bacterial abundance.
Protocol 2: Pre-Extraction Spike-In Control for Extraction Efficiency
Purpose: To quantitatively measure the loss of DNA during the extraction process.
- Prior to starting the extraction, add a known quantity (e.g., 10^4 copies) of an exogenous, non-competitor control DNA (e.g., from A. thaliana) to the sample lysis buffer.
- Proceed with the normal extraction protocol.
- Quantify the final eluate using qPCR with primers specific to the spike-in control.
- Calculation: % Recovery = (Copies recovered / Copies added) * 100. Efficiency >5-10% is often acceptable for complex samples like soil.
Visualizations
Diagram 1: Metagenomic DNA Extraction QC Workflow
(Diagram: Integrated QC Workflow for Metagenomic DNA)
Diagram 2: Troubleshooting Decision Tree Based on QC Results
(Diagram: Decision Tree for Interpreting QC Failures)
The Scientist's Toolkit: Essential Reagents & Materials
| Item | Function in QC | Key Considerations for Metagenomics |
|---|---|---|
| Fluorometric Dye Kits(e.g., Qubit dsDNA HS/BR) | Specific, dye-based quantification of dsDNA. Minimizes interference from RNA, ssDNA, and contaminants. | Use HS for low-biomass samples pre-library prep. BR for initial post-extraction yield. Always includes standards. |
| Inhibitor-Tolerant Polymerase(e.g., Platinum Taq, Phusion HP) | Enzymes for qPCR that withstand common environmental inhibitors (humic acids, phenols). | Essential for accurate amplification from soil/ sediment extracts. Often includes BSA. |
| Broad-Spectrum RNase A | Degrades contaminating RNA to prevent overestimation of DNA quality/yield on gels and fluorometry. | Use a DNAse-free, purified grade. Can be heat-inactivated if downstream RNA work is planned. |
| DNA Ladder (HMW)(e.g., Lambda HindIII) | Provides size reference on agarose gels to assess DNA integrity. | Confirm the presence of a distinct band >10 kb for optimal NGS library construction. |
| Exogenous Spike-in Control DNA(e.g., A. thaliana gBlock) | Added pre-extraction to quantitatively measure DNA recovery efficiency and identify inhibition. | Choose a sequence absent from your sample. Quantify precisely by digital PCR or spectrophotometry before use. |
| Inhibitor Removal Kit(e.g., silica-column or magnetic bead based) | Removes humic substances, polyphenols, and salts that inhibit enzymes. | Used as a clean-up step post-extraction if qPCR fails despite good fluorometry data. May cause some DNA loss. |
| Safe Nucleic Acid Stain(e.g., SYBR Safe, GelRed) | Intercalating dye for visualizing DNA on gels. Safer alternatives to ethidium bromide. | Compatible with blue-light transilluminators. Less mutagenic but requires similar handling precautions. |
Troubleshooting Guides & FAQs
Q1: Why do I observe significant genomic DNA contamination in my RNA extract from environmental samples, and how can I address it? A: gDNA contamination is common in co-extraction protocols due to shared chemical properties. To address this:
- In-Protocol DNase Treatment: Include an on-column or in-solution DNase I digestion step. For in-solution digestion after elution, use 2-5 U of DNase I per µg of total nucleic acid, incubate at 25-37°C for 15-30 minutes, then inactivate with EDTA.
- Verification: Always check RNA integrity and gDNA contamination by running an aliquot on an agarose gel (sharp rRNA bands, no high molecular weight smear) and by performing a no-reverse-transcriptase (No-RT) control in subsequent qPCR assays targeting a conserved gene (e.g., 16S rRNA gene). A Cq value >5 cycles later than the +RT control indicates acceptable gDNA removal.
Q2: My RNA yields from soil/sediment samples are consistently low. What are the key optimization points? A: Low yield often stems from inefficient cell lysis and RNA binding or RNase activity.
- Mechanical Lysis: Ensure rigorous bead-beating. Use a mixture of zirconia/silica beads (e.g., 0.1 mm and 0.5 mm) for 1-3 minutes at high speed. Keep samples cold during processing.
- Inhibit RNases: Pre-treat extraction reagents with diethyl pyrocarbonate (DEPC) or use certified RNase-free reagents and plastics. Include a potent RNase inhibitor (e.g., 1-2 U/µL) in the lysis buffer.
- Binding Conditions: For silica-column based methods, ensure the correct ethanol concentration (usually 70-75%) in the binding mixture. For low-biomass samples, consider carrier RNA (e.g., 1 µg of poly-A RNA) during precipitation or binding steps to improve recovery.
Q3: How do I choose between in-situ stabilization (e.g., RNAlater) and immediate flash-freezing for field sampling? A: The choice depends on sample type and logistics. See the comparison table below.
Table 1: Comparison of RNA Stabilization Methods for Field Sampling
| Method | Optimal Use Case | Key Advantage | Key Limitation | Typical Storage After Treatment |
|---|---|---|---|---|
| Flash-Freezing (LN₂/Dry Ice) | All sample types, if logistically feasible. | Instantaneous arrest of metabolic activity; gold standard. | Requires cryogens & continuous cold chain. | -80°C indefinitely. |
| Commercial Stabilizers (RNAlater) | Complex communities (gut, soil); when immediate freezing is impossible. | Permeates tissues, stabilizes RNA at ambient temp for 1 day, 4°C for 1 week. | Can be expensive for large volumes; may inhibit downstream enzymatic steps if not removed. | After 24h ambient, store at -80°C. |
| Ethanol-based Fixatives | High-microbial-biomass samples (e.g., fecal). | Cost-effective; good for morphology preservation. | May be less effective for RNA than dedicated reagents; requires precipitation cleanup. | -20°C to -80°C. |
Q4: What are the critical steps for successful co-extraction of DNA and RNA from the same sample aliquot? A: The sequential elution from a single column is a common approach. Key steps include:
- Simultaneous Lysis: Use a guanidinium thiocyanate-phenol-based lysis buffer (e.g., TRIzol or equivalent) to denature proteins and nucleases immediately.
- Phase Separation: Add chloroform, separate aqueous (RNA) and interphase/organic (DNA/protein) phases.
- RNA Recovery: Precipitate RNA from the aqueous phase with isopropanol, wash with 75% ethanol, and redissolve in RNase-free water or buffer.
- DNA Recovery: Precipitate DNA from the interphase and organic phase with ethanol, wash with sodium citrate in ethanol, and redissolve in TE buffer or water.
- Purification: Further purify both nucleic acid fractions using dedicated silica columns for highest quality.
Experimental Protocol: Sequential DNA/RNA Co-extraction from Soil Using a Phenol-Chloroform Method
- Materials: Liquid nitrogen, sterile pestle and mortar, PowerBead Tubes (0.1, 0.5 mm beads), Phenol:Chloroform:Isoamyl Alcohol (25:24:1, pH 4.5-5.0), TRIzol Reagent, Chloroform, Isopropanol, 75% Ethanol (RNase-free), DNase/RNase-free water, 3M Sodium Acetate (pH 5.2), Silica membrane spin columns.
- Procedure:
- Homogenization: Flash-freeze 0.5 g soil in LN₂, grind to fine powder. Transfer to a PowerBead tube containing 750 µL TRIzol and 250 µL sodium acetate (pH 5.0). Bead-beat for 2 min at 6 m/s.
- Phase Separation: Incubate 5 min at room temp. Add 200 µL chloroform, shake vigorously, centrifuge at 12,000 x g, 4°C, for 15 min. Transfer the upper aqueous phase (containing RNA) to a new tube.
- RNA Precipitation: To the aqueous phase, add 0.5x volume of RNase-free water and 0.7x volume of isopropanol. Mix and incubate at -20°C for 1 hour. Centrifuge at max speed, 4°C, for 30 min. Wash pellet with 75% ethanol, air-dry, and resuspend in 30 µL RNase-free water.
- DNA Recovery: To the interphase and organic phase, add 300 µL of 100% ethanol. Mix and incubate at RT for 3 min. Centrifuge at 2,000 x g, 4°C, for 5 min. Transfer supernatant to a new tube and precipitate DNA with 0.5 mL isopropanol. Wash pellet with ethanol containing sodium citrate, then with 70% ethanol. Resuspend DNA in TE buffer.
- Clean-up: Purify both RNA and DNA fractions using respective silica-column kits, including an on-column DNase step for RNA.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for RNA Preservation & Co-extraction
| Item | Function | Example/Critical Feature |
|---|---|---|
| RNase Inhibitors | Irreversibly bind to and inactivate RNases. | Recombinant RNasin or SUPERase•In. Critical for cell lysis steps. |
| Guanidinium Salts | Chaotropic agent; denatures proteins, inactivates RNases, promotes nucleic acid binding to silica. | Guanidine thiocyanate or hydrochloride in lysis buffers. |
| Acid-Phenol:Chloroform | Denatures and partitions proteins away from nucleic acids during phase separation. | pH 4.5-5.0 favors RNA partitioning to the aqueous phase. |
| Silica-Membrane Columns | Selective binding and purification of nucleic acids based on salt and ethanol conditions. | Dedicated RNA columns (bind >200 nt) and DNA columns. |
| Carrier RNA | Enhances recovery of low-concentration RNA during precipitation or column binding. | Poly-A RNA or glycogen (RNase-free). Avoids non-specific co-precipitation of inhibitors. |
| DNase I (RNase-free) | Digests contaminating genomic DNA post-extraction. | Required for metatranscriptomics. On-column digestion is preferred to avoid reintroducing RNases. |
| Stabilization Reagent | Penetrates tissue to inactivate RNases and stabilize RNA in situ prior to extraction. | RNAlater or similar. Allows for temporary non-freezer storage. |
| Bead Beating Matrix | Mechanically disrupts robust environmental matrices (soil, biofilms) and microbial cell walls. | Zirconia/silica beads of varying diameters (0.1, 0.5 mm) in a single tube. |
Visualizations
Title: Co-extraction Workflow for Metagenomics & Metatranscriptomics
Title: RNA Extraction Troubleshooting Decision Tree
Troubleshooting Guides & FAQs
Q1: My DNA yield from a soil metagenomic sample is consistently low with my current kit. Should I troubleshoot the protocol or switch kits? A: Begin with protocol optimization. Low yield is often due to inefficient cell lysis or inhibitor carryover. First, incorporate a mechanical lysis step (e.g., bead beating) if your kit uses only chemical lysis. Second, evaluate inhibitor removal by checking A260/A230 ratios; if low (<1.7), add a pre-wash step with a buffer like PBS or Sucrose-EDTA-Tris. Quantify results after each modification. Switching kits should be considered only if multiple optimization attempts fail and the cost of lost samples exceeds the cost of a new kit validated for your sample type.
Q2: I am getting high human host DNA contamination in my bacterial metagenomes from swab samples. Is this a kit limitation? A: Not necessarily. Most extraction kits co-extract DNA from all cells. Before switching, optimize by adding a selective lysis step. For bacterial enrichment, use a lysozyme/mutanolysin incubation step to pre-lyse bacterial cells, then degrade released DNA with a Benzonase treatment that cannot penetrate intact human cells, followed by a standard kit protocol. A kit switch is only warranted if you require a specialized commercial host depletion kit, which is significantly more expensive but may save time for high-volume processing.
Q3: My extracted DNA has poor purity (low A260/A280), failing downstream library prep. Should I change kits? A: First, optimize the wash steps. Ensure wash buffers contain the correct ethanol concentration and are not contaminated. Perform an additional wash or increase the dry time after washing to remove residual ethanol. If the issue persists, the silica membrane in the kit may be inadequate for your sample's inhibitor load. Switching to a kit with a different binding matrix (e.g., magnetic beads with tailored wash buffers) may be beneficial, as shown in the comparison table below.
Q4: The reproducibility of my extractions is poor. Is this a sign I need a new kit? A: Poor reproducibility is more often linked to protocol inconsistency than the kit itself. Standardize manual steps: use consistent homogenization times, precise incubation temperatures, and ensure column loads do not exceed capacity. If after rigorous standardization, inter-batch variability remains high, it may indicate kit quality control issues. Switching to a kit with a more robust, automated-friendly format (e.g., magnetic bead-based plates) could improve reproducibility for high-throughput studies.
Table 1: Cost-Benefit Analysis of Optimization vs. Switching
| Factor | Optimize Current Protocol | Switch to New Kit |
|---|---|---|
| Time Investment | High (iterative testing) | Medium (validation required) |
| Immediate Cost | Low (reagents only) | High (new kit purchase) |
| Risk of Failure | Moderate (incremental gains) | High (may not solve issue) |
| Long-Term Benefit | High (tailored method) | Medium (standardized) |
| Best For | Sample-specific issues, budget constraints | Obsolete kits, fundamental workflow flaws |
Table 2: Performance Metrics of Common DNA Extraction Approaches for Soil
| Method | Avg. Yield (ng/g) | A260/A280 | Cost per Sample | Protocol Time |
|---|---|---|---|---|
| Kit A (Silica Column) | 15 ± 5 | 1.78 ± 0.05 | $4.50 | 2.5 hrs |
| Kit A + Bead Beating | 42 ± 8 | 1.75 ± 0.08 | $5.00 | 3.0 hrs |
| Kit B (Magnetic Bead) | 38 ± 6 | 1.85 ± 0.03 | $7.00 | 2.0 hrs |
| Phenol-Chloroform | 55 ± 15 | 1.65 ± 0.10 | $1.50 | 4.5 hrs |
Experimental Protocols
Protocol: Optimization of a Silica-Column Kit for Inhibitor-Rich Soil
- Pre-Wash: Add 1g soil to 2 ml of Sucrose-EDTA-Tris buffer. Vortex, centrifuge (5000 x g, 5 min), discard supernatant.
- Enhanced Lysis: Resuspend pellet in kit lysis buffer. Add 0.3g of 0.1mm zirconia beads. Bead-beat at 6 m/s for 45 sec.
- Incubation: Heat at 70°C for 10 min. Centrifuge at 12,000 x g for 2 min.
- Binding: Transfer supernatant to a clean tube. Add 1.5x volume of kit binding buffer. Load onto column.
- Modified Wash: Perform first wash. For second wash, incubate buffer on column for 2 min before centrifugation.
- Elution: Elute with 50 µL of pre-warmed (70°C) nuclease-free water. Let column sit for 5 min before centrifugation.
Protocol: Comparative Validation for Kit Switching
- Sample Split: Divide a homogeneous, representative sample (e.g., soil slurry) into 6 aliquots.
- Parallel Processing: Extract 3 aliquots with the optimized current protocol and 3 with the new candidate kit.
- Quantification & Purity: Use fluorometry (e.g., Qubit) for yield and spectrophotometry (e.g., NanoDrop) for A260/A280/A230.
- Downstream QC: Perform qPCR on a conserved gene (e.g., 16S rRNA) to assess inhibitor presence and run a test library prep for NGS.
- Statistical Analysis: Use a t-test to compare yields and purity metrics. Proceed with switch only if the new kit shows significant (p<0.05) improvement in key metrics without increasing cost prohibitively.
Visualizations
Decision Flow: Optimize Protocol or Switch Kit
Paths from Sample Lysis to Improved DNA
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Metagenomic DNA Extraction |
|---|---|
| Zirconia/Silica Beads (0.1mm) | Mechanical shearing for robust cell wall lysis of tough microorganisms (e.g., Gram-positives) in bead beating. |
| Inhibitor Removal Technology (IRT) Buffers | Commercial or custom buffers containing compounds that sequester humic acids, polyphenols, and other common environmental inhibitors. |
| Lysozyme & Mutanolysin | Enzymes that hydrolyze bacterial peptidoglycan cell walls, enabling selective lysis or enhanced lysis efficiency. |
| Benzonase Nuclease | Degrades free DNA and RNA in samples; useful for host depletion strategies when used with selective lysis. |
| Sucrose-EDTA-Tris Buffer | A pre-wash buffer for soil/sediment that helps remove soluble inhibitors prior to the main lysis step. |
| SPRI Magnetic Beads | Size-selective magnetic beads for DNA cleanup and size selection post-extraction, improving library prep success. |
| PCR Inhibitor Spin Columns | Specialized silica columns designed to bind and remove specific inhibitor classes from difficult samples. |
| Guanidine Hydrochloride | A potent chaotropic salt used in lysis buffers to denature proteins and facilitate DNA binding to silica. |
Benchmarking Performance: How to Validate and Compare DNA Extraction Methods
Technical Support Center
Troubleshooting Guides & FAQs
Q1: My DNA yield from a soil metagenomic sample is consistently low. What are the primary causes and solutions?
A: Low yield is commonly caused by inefficient cell lysis or DNA retention on soil particles.
- Check: Lysis Method. Silica-based soils require mechanical lysis (e.g., bead beating). Use a combination of chemical, enzymatic, and mechanical lysis for robust communities.
- Protocol: Increase bead-beating time (adjust from 30s to 2-3 min in cycles with cooling) or use smaller/ more abrasive beads (e.g., 0.1mm zirconia).
- Check: Inhibitor Carryover. Humic acids inhibit downstream reactions. Use inhibitor removal columns specifically designed for environmental samples or add polyvinylpolypyrrolidone (PVPP) to the lysis buffer.
- Quantify: Always use fluorescence-based assays (e.g., Qubit) over UV absorbance for accurate yield measurement in the presence of contaminants.
Q2: My extracted DNA has high 260/230 and 260/280 ratios, but PCR/sequencing fails. What unseen contaminants should I suspect?
A: Acceptable ratios (1.8-2.0 and >2.0 respectively) do not guarantee functional purity. Persistent failure suggests carryover of enzymatic inhibitors.
- Troubleshoot: Perform a 1:5 and 1:10 dilution of your DNA in a standard PCR. If the diluted samples work, inhibitor presence is confirmed.
- Solution: Perform a post-extraction cleanup using silica membrane columns with an added wash step of 5mM ammonium acetate in 80% ethanol to remove salts and organics. Alternatively, use gel electrophoresis or Fragment Analyzer to check for RNA contamination, which inflates 260/280.
- Protocol - Post-Extraction Cleanup: Bind DNA to a silica column, wash with 80% ethanol, perform an additional wash with 5mM ammonium acetate in 80% ethanol, then elute.
Q3: My fragment size analysis shows excessive shearing (<500bp). How can I optimize for longer fragments?
A: Excessive shearing occurs during cell lysis or subsequent handling.
- Optimize Lysis: Reduce mechanical lysis intensity. For bead beating, try shorter pulses (e.g., 3 x 20s with 60s cooling on ice).
- Handle Gently: Avoid vortexing after lysis. Use wide-bore pipette tips for all transfers post-lysis. Do not vigorously mix binding solutions; invert tubes gently.
- Elution Buffer: Always elute in pre-warmed (55°C) low-EDTA TE buffer or nuclease-free water, and let it sit on the membrane for 2-5 minutes before centrifugation.
- Storage: Store DNA at -80°C in aliquots to avoid freeze-thaw shearing.
Q4: My sequencing results show poor representation of Gram-positive bacteria. How can I improve lysis efficiency for robust cells?
A: Gram-positive bacteria have thick peptidoglycan layers resistant to standard lysis.
- Protocol Enhancement: Incorporate a pre-lysis enzymatic step. Resuspend the pellet in a lysozyme (20 mg/ml) and mutanolysin (5 U/ml) solution in TE buffer. Incubate at 37°C for 60 minutes before proceeding to mechanical lysis.
- Lysis Buffer: Use a specialized lysis buffer containing CTAB and proteinase K for difficult samples.
- Mechanical Lysis: Ensure your bead-beating protocol includes a mix of bead sizes (e.g., 0.1mm and 0.5mm) to maximize disruption efficiency.
Q5: How do I validate that my extraction method does not introduce bias in microbial community representation?
A: Use a mock microbial community standard comprising known, quantitated strains from a diverse range of taxa.
- Experiment: Extract DNA from the mock community using your protocol in parallel with a published, low-bias method.
- Analysis: Perform 16S rRNA gene sequencing (for bacteria/archaea) or shotgun sequencing. Compare the relative abundances recovered by each method to the known input ratios.
- Metric: Calculate the bias as (Observed Abundance / Expected Abundance). A perfect method gives a value of 1 for all members.
Data Presentation: Validation Metrics for Common Kits
Table 1: Comparison of Commercial Metagenomic DNA Extraction Kits
| Kit Name | Optimal Sample Type | Avg. Yield (Soil) | Typical Fragment Size | Inhibitor Removal | Bias Assessment (Gram+ vs. Gram-) |
|---|---|---|---|---|---|
| PowerSoil Pro Kit | Difficult, inhibitor-rich | 0.5 - 5 µg/g | 10 - 20 kb | Excellent | Moderate (Improved with longer beating) |
| DNeasy PowerLyzer | Mechanically tough | 1 - 10 µg/g | 5 - 15 kb | Very Good | Low (Robust mechanical lysis) |
| FastDNA SPIN Kit | Broad environmental | 2 - 15 µg/g | 1 - 10 kb | Good | High bias against robust cells |
| MetaPolyzyme Method | Human gut, Gram+ rich | 0.1 - 2 µg/g | 20 - 50 kb | Moderate | Very Low (Enzymatic lysis focus) |
Note: Yield is highly sample-dependent. Values are indicative ranges from recent literature (2023-2024).
Experimental Protocols
Protocol 1: Comprehensive Extraction from Soil with Inhibitor Removal
Title: Optimized Soil Metagenomic DNA Extraction Protocol.
Materials: Soil sample, PowerSoil Pro Kit (QIAGEN), 0.1mm zirconia beads, bead beater, microcentrifuge, 55°C water bath.
Steps:
- Homogenize: Weigh 0.25g of soil into a PowerBead Tube.
- Lysis: Add 60µl of Solution C1 (inhibitor removal reagent). Secure tubes horizontally on bead beater.
- Mechanical Disruption: Beat at 6.0 m/s for 45 seconds. Incubate on ice for 2 minutes. Repeat twice (total 3 cycles).
- Binding: Centrifuge. Transfer supernatant to a clean tube. Add 250µl of Solution C2 and vortex for 5s. Incubate at 4°C for 5 min. Centrifuge. Transfer supernatant to a new tube.
- Inhibitor Precipitation: Add 200µl of Solution C3, vortex, incubate at 4°C for 5 min. Centrifuge.
- DNA Binding: Transfer supernatant to an MB Spin Column. Centrifuge. Discard flow-through.
- Wash: Add 500µl of Solution C4. Centrifuge. Discard flow-through.
- Dry: Centrifuge empty column for 1 minute.
- Elute: Place column in clean tube. Add 50µl of pre-warmed (55°C) Solution C6 (10mM Tris). Incubate at room temp for 3 min. Centrifuge for 1 min.
Protocol 2: Validation Using a Mock Microbial Community
Title: Protocol for Assessing Extraction Bias with a Mock Community.
Materials: ZymoBIOMICS Microbial Community Standard (cat# D6300), extraction kit/test protocol, Qubit fluorometer, sequencing platform.
Steps:
- Sample Prep: Resuspend the mock community pellet according to manufacturer instructions. Split into aliquots for extraction replicates.
- Parallel Extraction: Extract DNA from the mock community using your test protocol and a reference protocol (e.g., a published, phenol-chloroform based method) in triplicate.
- Quantification: Quantify DNA yield using a fluorescence assay (Qubit dsDNA HS).
- Library Prep & Sequencing: Prepare 16S rRNA gene amplicon libraries (targeting V3-V4) for all extracts using the same master mix and conditions. Sequence on an Illumina MiSeq with sufficient depth (>100,000 reads/sample).
- Bioinformatic Analysis: Process reads through a standard pipeline (DADA2, QIIME2). Assign taxonomy using a trained classifier.
- Bias Calculation: For each known member i of the mock community, calculate: Bias Index = (Relative Abundance in Test Extract) / (Relative Abundance in Reference Extract). Plot results.
Mandatory Visualizations
Diagram 1: Metagenomic DNA Extraction & Validation Workflow
Diagram 2: Common Issues & Mitigation Pathways
The Scientist's Toolkit
Table 2: Essential Research Reagents & Materials for Metagenomic DNA Extraction
| Item | Function & Rationale |
|---|---|
| Zirconia/Silica Beads (0.1mm & 0.5mm) | Provides mechanical shearing for robust cell wall disruption. A mix of sizes increases lysis efficiency across diverse cell types. |
| Inhibitor Removal Technology (IRT) / PVPP | Binds to humic acids, polyphenols, and other common environmental inhibitors that co-precipitate with DNA and hinder downstream applications. |
| Lysozyme & Mutanolysin | Enzymes that hydrolyze peptidoglycan layers, specifically improving lysis of Gram-positive bacterial cells which are often underrepresented. |
| CTAB (Cetyltrimethylammonium bromide) | A cationic detergent effective in lysing cells and separating DNA from polysaccharides and other contaminants in complex samples. |
| DNA Binding Silica Membranes/ Magnetic Beads | Selective binding of DNA in high-salt conditions, allowing for efficient washing and purification away from proteins, salts, and other impurities. |
| Mock Microbial Community Standard | A defined mix of microbial genomes with known abundances. The critical standard for validating extraction bias and benchmarking protocol performance. |
| Fluorometric DNA Assay (e.g., Qubit dsDNA HS) | Provides accurate DNA quantification by specifically binding double-stranded DNA, unaffected by common contaminants that skew UV absorbance readings. |
| Fragment Analyzer / Bioanalyzer | Provides precise sizing and qualitative assessment of extracted DNA, critical for ensuring fragment length is suitable for long-read or short-read sequencing. |
Inter-laboratory Comparisons and Standard Reference Materials (e.g., ZymoBIOMICS Mock Communities)
Technical Support Center
FAQs & Troubleshooting Guides
Q1: Our extracted DNA yield from the ZymoBIOMICS Gut Microbiome Standard is consistently lower than the expected range provided in the datasheet. What are the potential causes? A: Low DNA yield can result from several protocol deviations. First, ensure the mock community pellet is fully resuspended before extraction—vortex for 5 minutes, not just briefly. Second, check the lysis conditions; mechanical disruption (e.g., bead beating) is mandatory for robust Gram-positive bacterial lysis. Insufficient bead beating time or speed will reduce yield. Third, confirm that elution buffer is pre-warmed (e.g., 55°C) and that you are eluting in a minimal volume (e.g., 50-100 µL) directly onto the silica membrane. Do not use water for elution. Finally, verify the quantification method; fluorometric assays (Qubit) are strongly recommended over absorbance (NanoDrop) for accuracy.
Q2: We observe significant deviations from the expected taxonomic profile in our sequencing data after processing a mock community. How should we troubleshoot? A: This indicates bias introduced during wet-lab or bioinformatics steps. Follow this systematic approach:
- Wet-Lab Verification: Re-run the extraction and quantify with Qubit. Ensure you are loading the recommended DNA amount into your library prep. Cross-check every reagent batch and use freshly aliquoted, PCR-grade water.
- Control Check: Include a "no-template" PCR/Library prep control to rule out contamination.
- Bioinformatics Audit: Use the exact, version-matched reference database provided by Zymo. Re-analyze raw FASTQ files with a standard, minimal bioinformatics pipeline (e.g., direct read mapping with Bowtie2 or Kraken2) to see if bias persists before using your complex metagenomic pipeline.
- Bias Localization: If the bias is consistent across replicates, it likely originates in the DNA extraction or library preparation step. Compare your protocol's lysis conditions to the manufacturer's validated protocol.
Q3: How should we incorporate mock communities into our experimental design for a thesis on DNA extraction methods? A: Mock communities serve as critical process controls. For a comparative extraction methods thesis:
- Placement: Include them at the start of every extraction batch.
- Replication: Process each mock community in triplicate for each extraction method being evaluated.
- Purpose: They allow you to quantify method-specific biases in yield, community composition, and even integrity (via QC like Qubit/ Fragment Analyzer). This data is essential for justifying the selection of an optimal extraction method for your specific sample matrix (e.g., soil, stool).
Q4: What are the key metrics to report when publishing inter-laboratory comparison data using mock communities? A: Transparency is key. Report the data summarized in the table below:
Table 1: Key Quantitative Metrics for Reporting Inter-laboratory Comparisons Using Mock Communities
| Metric | How to Calculate/Report | Purpose in Thesis Context |
|---|---|---|
| DNA Yield | ng of DNA per sample volume/weight. Report mean ± SD. | Compare extraction efficiency across methods. |
| Purity (A260/A280) | Absorbance ratio. Ideal range ~1.8-2.0. | Indicate presence of co-extracted contaminants that inhibit downstream steps. |
| Taxonomic Bias | Relative abundance of each known species vs. expected truth. Use bar charts. | Identify which methods over/under-represent specific taxa (e.g., Gram-positives). |
| Alpha Diversity Metrics | Observed Species, Shannon Index on known composition. | Assess if a method "recovers" the expected richness and evenness. |
| Inter-lab CV | Coefficient of Variation for abundance of key taxa across participating labs. | Demonstrate the reproducibility afforded by a standardized protocol. |
| Limit of Detection | Lowest input cell quantity from which a taxon is reliably detected. | Show sensitivity of the extraction and sequencing pipeline. |
Experimental Protocols
Protocol 1: Validated DNA Extraction from ZymoBIOMICS Mock Communities using a Bead-Beating Method This protocol is adapted for a thesis comparing mechanical lysis efficiency.
Materials: ZymoBIOMICS Microbial Community Standard (D6300), ZymoBIOMICS DNA Miniprep Kit (D4300), sterile 2.0 mL screw-cap tubes, 0.1 & 0.5mm zirconia/silica beads, vortex adapter, microcentrifuge, fluorometer (Qubit).
Methodology:
- Resuspension: Thaw the mock community vial on ice. Vortex vigorously for 5 minutes to ensure a homogeneous suspension.
- Lysis: Transfer 200 µL of the suspension to a bead tube provided in the kit. Add 750 µL of ZymoBIOMICS Lysis Solution.
- Mechanical Disruption: Secure tubes in a vortex adapter. Vortex at maximum speed for 25 minutes. Thesis Note: This step is a key variable; compare times of 5, 15, and 25 mins.
- DNA Binding & Wash: Centrifuge at 10,000 x g for 1 min. Transfer supernatant to a Zymo-Spin III-F filter. Centrifuge. Wash with 400 µL of DNA Wash Buffer. Centrifuge.
- Elution: Apply 50 µL of pre-heated (55°C) DNA Elution Buffer directly to the filter matrix. Incubate at room temp for 2 minutes. Centrifuge at full speed for 1 minute to elute.
- Quantification: Quantify DNA using the Qubit dsDNA HS Assay. Avoid absorbance methods for accurate yield assessment.
Protocol 2: Bioinformatics Validation of Extraction Fidelity Protocol for analyzing sequencing output from mock communities.
Materials: Raw paired-end FASTQ files, ZymoBIOMICS reference sequences (download from https://zenodo.org/record/8017460), Bowtie2, SAMtools.
Methodology:
- Index Reference: Build a Bowtie2 index from the ZymoBIOMICS reference FASTA file:
bowtie2-build reference.fasta zymo_index - Read Mapping: Map sample reads to the index:
bowtie2 -x zymo_index -1 sample_R1.fastq -2 sample_R2.fastq -S output.sam --no-unal - Processing: Convert SAM to BAM, sort, and index:
samtools view -bS output.sam | samtools sort -o sorted.bam && samtools index sorted.bam - Coverage Calculation: Generate per-taxon coverage:
samtools depth sorted.bam > coverage.txt - Analysis: Calculate relative abundance from read counts mapped to each reference genome. Compare to the known theoretical composition.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Controlled Metagenomic Studies
| Item | Function & Rationale |
|---|---|
| ZymoBIOMICS Microbial Community Standard (D6300) | Defined synthetic mock community. Serves as an absolute control for DNA extraction, library prep, and bioinformatics pipeline validation. |
| ZymoBIOMICS Spike-in Control (D6320) | Defined set of odd-ratio microbes. Added to a native sample to track and correct for technical bias across the entire workflow. |
| ZymoBIOMICS DNA Miniprep Kit (D4300) | Optimized kit for mock communities. Includes inhibitors to mimic difficult samples and standardized bead-beating tubes. |
| Qubit dsDNA HS Assay Kit | Fluorometric quantification. Essential for accurate measurement of low-concentration, potentially contaminated metagenomic DNA, unlike UV absorbance. |
| PCR-Grade Water, nuclease-free | Used for all critical dilutions and reconstitutions. Prevents contamination from nucleases or microbial DNA present in lab-purified water. |
| Certified Low-Binding DNA LoBind Tubes | Minimizes DNA adhesion to tube walls during extraction and library preparation, critical for low-biomass samples. |
Workflow & Relationship Diagrams
Title: Thesis Workflow for Evaluating DNA Extraction Methods Using Mock Controls
Title: Sources of Bias from Truth to Observed Taxonomic Profile
Troubleshooting Guide & FAQs
Q1: During bioinformatic analysis, my alpha diversity metrics (e.g., Shannon Index) show unexpectedly low values after using a specific DNA extraction kit. What could be the cause and how can I troubleshoot this? A: Low alpha diversity often indicates poor lysis of tough-to-lyse microorganisms (e.g., Gram-positive bacteria, spores), leading to biased community representation.
- Troubleshooting Steps:
- Verify Protocol: Ensure you included a mechanical lysis step (e.g., bead beating) as per the kit's instructions for environmental/metagenomic samples. Omission is a common error.
- Increase Bead-Beating Time: If protocol included bead beating, increase the homogenization time by 30-second increments in a new sample batch. Over-beating can shear DNA, but under-beating is a more frequent issue for diversity.
- Incorporate a Pre-treatment: For soil or stool samples, add a pre-incubation step with a chelating agent (e.g., 10mM EDTA) or a lysozyme incubation (30 min, 37°C) prior to the kit's standard protocol.
- Positive Control: Spike a known quantity of a hard-to-lyse organism (e.g., Bacillus subtilis spores) into your sample and re-extract. Quantify recovery via qPCR to confirm lysis efficiency.
Q2: My beta diversity plots (PCoA based on Bray-Curtis) show high technical replicate dispersion, suggesting high variability. How can I improve consistency? A: High technical variability usually stems from inconsistencies in the initial steps of biomass handling and lysis.
- Troubleshooting Steps:
- Standardize Homogenization: Ensure the original sample is thoroughly homogenized before subsampling. For solid samples, use a consistent, powerful homogenizer.
- Check Bead Beating Consistency: If using a vortex adapter for bead beating, ensure tubes are securely fastened in a consistent orientation. Consider switching to a high-throughput homogenizer (e.g., Mo Bio Homogenizer, MP Biomedicals FastPrep) for more uniform mechanical force.
- Monitor Inhibitor Carryover: Inconsistent inhibitor removal (humics, bile salts) can cause variable PCR amplification. Perform a 1:10 dilution of your extracted DNA and re-run PCR. If clustering improves, re-optimize the inhibitor removal steps or use a kit with more stringent wash buffers.
- Elution Volume Precision: Always elute in a precise, pre-warmed elution buffer. Inconsistent elution volume dramatically alters concentration and downstream library prep consistency.
Q3: I am comparing two extraction methods. My negative control (blank) shows non-negligible reads after sequencing, complicating beta diversity interpretations. How should I proceed? A: Contamination in controls invalidates diversity metrics. This is a critical issue requiring systematic decontamination.
- Troubleshooting Steps:
- Identify Contaminant Source: BLAST the predominant sequences in the blank against known contaminants (e.g., Pseudomonas, Comamonadaceae, Burkholderia). These often originate from kit reagents or lab environments.
- UV-Irradiate Consumables: Prior to extraction, expose all plasticware (tips, tubes) and solutions (PCR water, buffers not provided with kit) to UV radiation in a crosslinker for 20 minutes.
- Use Dedicated Kits & Areas: Designate separate extraction kits and pipettes for pre- and post-PCR work. Perform extractions in a dedicated, clean hood if possible.
- Statistical Correction: If contamination is minimal and uniform, use bioinformatic tools like
decontam(R package) infrequencyorprevalencemode to identify and remove contaminant sequences from your feature table BEFORE calculating diversity metrics.
Table 1: Impact of Lysis Stringency on Alpha Diversity Metrics (Simulated Soil Community)
| Extraction Protocol Modifier | Shannon Index (Mean ± SD) | Observed ASVs (Mean ± SD) | Pielou's Evenness (Mean ± SD) |
|---|---|---|---|
| Kit Protocol Only (No bead beating) | 4.2 ± 0.3 | 350 ± 25 | 0.78 ± 0.04 |
| Kit Protocol + Standard Bead Beating (90s) | 6.8 ± 0.2 | 620 ± 30 | 0.92 ± 0.02 |
| Kit Protocol + Extended Bead Beating (180s) | 6.5 ± 0.4 | 590 ± 45 | 0.88 ± 0.03 |
| Phenol-Chloroform + Bead Beating (Reference) | 7.1 ± 0.3 | 655 ± 35 | 0.93 ± 0.01 |
Table 2: Effect of Inhibitor Removal on Beta Diversity Distance (Bray-Curtis) Between Technical Replicates
| Sample Type | Extraction Kit (With Inhibitor Removal) | Mean Distance | Extraction Kit (Minimal Wash Steps) | Mean Distance |
|---|---|---|---|---|
| Fecal | Kit A (Silica + Inhibitor Wash) | 0.12 ± 0.03 | Kit B (Silica only) | 0.31 ± 0.07 |
| Soil (High Humics) | Kit C (CTAB + Silica) | 0.15 ± 0.04 | Kit A (Silica + Inhibitor Wash) | 0.24 ± 0.05 |
Experimental Protocols
Protocol 1: Evaluating Lysis Efficiency on Alpha Diversity Objective: Systematically test the impact of mechanical lysis duration on recovery of microbial diversity.
- Sample Preparation: Aliquot 0.25g of a homogenized, standardized soil sample (e.g., ZymoBIOMICS Soil Reference) into ten 2mL screw-cap tubes.
- Lysis Variation: To all tubes, add the recommended lysis buffer from a commercial kit (e.g., DNeasy PowerSoil Pro Kit). Process five tubes with bead beating at 4 m/s for 60s (standard). Process the other five for 180s (extended).
- DNA Extraction: Complete the remaining extraction steps (inhibition removal, binding, washing, elution) identically for all tubes as per the kit manual. Elute in 50µL.
- Library Prep & Sequencing: Quantify DNA with Qubit dsDNA HS Assay. For each extract, prepare 16S rRNA gene amplicon libraries (V4 region) using the same master mix and cycle count. Pool libraries equimolarly and sequence on an Illumina MiSeq (2x250bp).
- Bioinformatic Analysis: Process reads through DADA2 pipeline to generate ASV tables. Calculate alpha diversity metrics (Shannon, Observed ASVs, Pielou's Evenness) in QIIME2.
- Statistical Analysis: Compare alpha diversity metrics between standard and extended beating groups using a non-parametric Mann-Whitney U test.
Protocol 2: Assessing Technical Reproducibility for Beta Diversity Objective: Quantify the impact of extraction protocol consistency on beta dispersion.
- Replicate Design: From a single, well-homogenized source (e.g., frozen fecal aliquot), take ten 200mg subsamples.
- Extraction: Randomly assign five subsamples to "Technician A" and five to "Technician B." Each technician performs the full extraction protocol (e.g., MagAttract PowerSoil DNA Kit) independently but identically.
- Downstream Processing: All ten extracts proceed through independent but standardized library preparation, sequencing, and ASV table generation (as in Protocol 1).
- Beta Diversity Calculation: Compute the Bray-Curtis dissimilarity matrix between all samples in QIIME2.
- Visualization & Analysis: Generate a PCoA plot. Calculate the mean distance of replicates to their respective group centroid (i.e., beta dispersion) using the
betadisperfunction in R (veganpackage). Compare dispersion between Technician groups via PERMDISP.
Visualizations
Diagram Title: DNA Extraction Workflow Highlighting Critical Lysis Step
Diagram Title: Troubleshooting Low or Inconsistent Diversity Metrics
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Evaluating Extraction Impact on Diversity
| Item | Function in Context of Diversity Analysis |
|---|---|
| Mechanical Lysis Beads (e.g., 0.1mm silica/zirconia) | Essential for breaking tough cell walls. Inconsistent bead size/shar causes variable lysis, impacting alpha diversity. |
| Standardized Mock Microbial Communities (e.g., ZymoBIOMICS) | Ground-truth positive control with known composition. Critical for benchmarking alpha/beta diversity results from different extraction methods. |
| Inhibitor Removal Buffers (e.g., guanidine thiocyanate, PTB) | Removes humics, polyphenols, and salts that inhibit PCR. Inefficient removal increases stochasticity, inflating beta dispersion. |
| DNA Binding Silica Membranes/Magnetic Beads | Purifies DNA from lysate. Binding capacity and purity affect yield and downstream sequencing library uniformity. |
| DNase-/RNase-Free Water with UV Treatment | Used for final elution and reagent prep. Minimizes contaminating environmental DNA that skews rare biosphere detection. |
| High-Sensitivity DNA Quantification Assay (e.g., Qubit dsDNA HS) | Accurate quantification is vital for equal library pooling. Fluorometric assays avoid contamination from RNA/proteins unlike absorbance. |
| PCR Enzyme Mix with Proofreading | For 16S/ITS amplicon or shotgun library prep. High-fidelity polymerase reduces chimera formation, preserving true beta diversity signals. |
Troubleshooting Guides & FAQs
Q1: My 16S rRNA amplicon sequencing results show very low diversity and high abundance of a single bacterial genus. What could be the cause? A: This is often due to primer bias. The universal primers used (e.g., 27F/1492R for the full gene or V4 region primers) may have mismatches for your specific sample matrix. Verify primer sequences against updated databases like SILVA. Consider using a primer cocktail (multiple forward/reverse primers) to broaden coverage. Also, check for PCR over-amplification; reduce cycle number and use a high-fidelity polymerase.
Q2: During shotgun metagenomic DNA extraction, I'm getting sheared DNA with fragments mostly below 1kb, impacting assembly. How can I improve integrity? A: This indicates mechanical or chemical degradation. Implement gentle lysis: for tough Gram-positive bacteria, use enzymatic lysis (lysozyme, mutanolysin) at 37°C for 60 min before any bead-beating. If bead-beating is essential, use larger (e.g., 0.5mm) beads and shorter pulse times (3x 30s pulses with cooling). Include an RNase A step to reduce viscosity before purification. Use silica-membrane columns with a >10kb cutoff or switch to magnetic bead-based size selection.
Q3: My viral metagenomics (virome) prep is contaminated with abundant host and bacterial DNA. How do I enrich for viral particles? A: Implement sequential filtration and density centrifugation. Pass the sample through a 0.22µm PES filter to remove bacteria/cells. Follow with a 0.1µm filter to capture larger viruses. For further purification, use a Benzonase nuclease treatment (37°C, 30-60 min) to digest unprotected nucleic acid (from lysed cells), which does not penetrate viral capsids. A final step of CsCl or sucrose density gradient ultracentrifugation can effectively isolate viral particles based on buoyant density.
Q5: How do I choose between 16S rRNA and shotgun metagenomics for a gut microbiome study aiming to find therapeutic targets? A: Base the choice on your resolution and functional insight needs. Use 16S rRNA for cost-effective, high-depth profiling of bacterial community composition and broad taxonomic shifts across many samples. Use shotgun metagenomics when you need species- or strain-level identification, need to profile non-bacterial kingdoms (archaea, fungi, viruses), or require information on functional genes and metabolic pathways. A tiered approach (16S for screening, shotgun for deep dive on select samples) is common.
Table 1: Comparison of Key Metagenomic Extraction Method Characteristics
| Feature | 16S rRNA Amplicon Sequencing | Shotgun Metagenomics | Viral Metagenomics (Virome) |
|---|---|---|---|
| Primary Target | Hypervariable regions of prokaryotic 16S rRNA gene | All genomic DNA in sample | Viral nucleic acids (DNA, RNA, or both) |
| Typical DNA Input | 1-10 ng | 1-100 ng (varies by kit) | 0.1-10 ng (often requires amplification) |
| Average Read Length | 250-600 bp (Illumina MiSeq) | 150 bp - 10+ kb (Illumina vs. PacBio) | 150-300 bp (Illumina) |
| Sequencing Depth Needed | 10,000-50,000 reads/sample | 10-50 million reads/sample | 5-100 million reads/sample |
| Cost per Sample | $20 - $100 | $100 - $1000+ | $150 - $800 |
| Key Advantage | High-depth taxonomic profiling of bacteria/archaea; cost-effective for many samples. | Captures all genetic material; enables functional pathway analysis & broader kingdom ID. | Specifically profiles viral community; can discover novel viruses. |
| Main Limitation | Primer bias; limited to bacteria/archaea; no functional data. | Host DNA contamination; computationally intensive; higher cost. | Extremely sensitive to contamination; complex wet-lab protocol. |
| Best for Applications | Microbial community surveys, ecological studies, initial diagnostics. | Functional potential discovery, pathogen detection, multi-kingdom analysis. | Viral discovery, viral ecology, phage therapy, viral epidemiology. |
Table 2: Essential Research Reagent Solutions Toolkit
| Reagent / Material | Function | Primary Application |
|---|---|---|
| Lytic Enzymes (Lysozyme, Mutanolysin) | Breaks down bacterial cell walls (peptidoglycan). | Gentle lysis for Gram-positive bacteria in shotgun/16S prep. |
| Proteinase K | Degrades proteins and inactivates nucleases. | Standard step in most DNA extraction protocols post-lysis. |
| Benzonase Nuclease | Degrades all forms of DNA and RNA (linear, circular, chromosomal). | Removal of free nucleic acids outside viral capsids in virome prep. |
| Phi29 DNA Polymerase | Used in Multiple Displacement Amplification (MDA). | Whole-genome amplification of low-input viral or microbial DNA. |
| Size-selection Beads (SPRI) | Selective binding of DNA by size (PEG/NaCl solution). | Removing short fragments, primer dimers, and selecting insert size. |
| CsCl / Sucrose | Forms density gradient for ultracentrifugation. | Purification and concentration of viral particles based on buoyant density. |
| PMSF (Protease Inhibitor) | Inhibits serine proteases. | Preserves proteins, including viral capsids, during sample processing. |
| DNase I (RNase-free) | Degrades single/double-stranded DNA. | Optional for removing host DNA in virome prep after lysis of non-viral particles. |
Experimental Protocols
Protocol 1: Standardized Shotgun Metagenomic DNA Extraction from Stool (Modified from the IHMS SOP)
- Homogenization: Weigh 100-200 mg of stool. Suspend in 1 mL of lysis buffer (500 mM NaCl, 50 mM Tris-HCl pH 8, 50 mM EDTA, 4% SDS).
- Mechanical Lysis: Add 0.5 g of 0.1mm zirconia/silica beads. Bead-beat at high speed for 2-3 minutes.
- Incubation: Incubate at 95°C for 5 minutes to further lyse cells and inactivate nucleases.
- Precipitation: Add 260 µL of 10 M ammonium acetate, mix, and incubate on ice for 5 minutes. Centrifuge at 16,000 x g for 10 min.
- Binding: Transfer supernatant to a new tube. Add 0.7 volumes of isopropanol, mix. Load onto a silica-membrane column.
- Wash & Elute: Wash twice with 70% ethanol. Elute DNA in 50-100 µL of TE buffer or nuclease-free water.
- QC: Assess quantity (Qubit dsDNA HS assay) and quality (Fragment Analyzer or Bioanalyzer for size distribution).
Protocol 2: Viral Particle Enrichment and DNA Extraction from Serum for Virome Analysis
- Clarification: Centrifuge 1 mL serum at 5,000 x g for 10 min at 4°C to remove cells/debris.
- Filtration: Pass supernatant through a 0.22 µm syringe filter, followed by a 0.1 µm filter.
- Nuclease Treatment: Add MgCl₂ to 2 mM final concentration. Add 2-5 µL Benzonase nuclease (25 U/µL). Incubate at 37°C for 60 min.
- Viral Lysis & DNA Extraction: Add Proteinase K and SDS (final 0.5%). Incubate at 56°C for 30 min.
- Purification: Perform phenol-chloroform-isoamyl alcohol extraction, followed by ethanol precipitation. Alternatively, use a commercial column-based kit designed for low-copy-number DNA.
- Amplification (if needed): Perform Multiple Displacement Amplification (MDA) using a phi29-based kit (e.g., REPLI-g) following manufacturer's guidelines for low-input DNA.
Workflow Diagrams
Title: Decision Workflow for Metagenomic Method Selection
Title: Viral Metagenomics Sample Processing Workflow
Title: Lysis Method Trade-offs for DNA Yield & Integrity
FAQs & Troubleshooting Guide
Q1: Why does my metagenomic DNA extraction yield from a gut microbiota sample vary drastically between bead-beating and enzymatic lysis protocols, and which is more relevant for antibiotic resistance gene discovery? A: Yield variation stems from differential lysis efficiency for Gram-positive vs. Gram-negative bacteria. Bead-beating is harsher, breaking tough cell walls (e.g., Firmicutes), while enzymatic lysis is gentler. For antibiotic resistance gene discovery, under-representing Gram-positives can miss key genes. A 2019 study in Nature Communications compared four extraction kits on stool samples. The harsher mechanical method revealed 33% more putative antibiotic resistance genes compared to a gentle kit, dramatically altering the inferred resistome profile and potential drug targets.
Q2: We observed contradictory microbial signatures for colorectal cancer (CRC) biomarker discovery when comparing two studies. Could DNA extraction be a factor? A: Yes. Inconsistent extraction can skew microbial community representation. A 2020 benchmark study in Microbiome analyzed five extraction methods from identical CRC biopsy samples. Methods favoring certain bacterial lineages led to different candidate diagnostic biomarkers. For instance, one method elevated Fusobacterium signals, while another amplified Prevotella. This directly impacts the development of a consensus, non-invasive diagnostic test.
Q3: After switching to a new soil DNA kit for natural product discovery, we no longer detect Actinobacteria sequences. What went wrong? A: Actinobacteria have thick, mycelial cell walls resistant to gentle lysis. Your new kit may lack sufficient mechanical disruption. Research in The ISME Journal (2021) demonstrated that a protocol incorporating extended bead-beating and a heated lysis step increased Actinobacteria detection by over 40% in complex soils. Since Actinobacteria are a prime source of novel drug-like molecules, this extraction shift could cause you to miss a critical biosynthetic gene cluster.
Q4: My viral enrichment protocol for phage-derived therapeutic proteins is co-extracting high levels of host DNA, hampering sequencing efficiency. How can I improve specificity? A: This is common in virome studies. The issue is often insufficient removal of free host DNA and non-viral particles. Incorporate a step of 0.22 µm filtration followed by DNase treatment (benzonase) on the filtrate to digest unprotected nucleic acids before viral particle lysis. A 2022 Cell Reports Methods protocol showed this combination reduced host DNA contamination from >80% to <15% in sputum samples, enabling efficient sequencing of viral genomes for therapeutic discovery.
Q5: Why do my extracted DNA fragments from formalin-fixed paraffin-embedded (FFPE) tissue for microbiome-based oncology biomarkers show extreme fragmentation and low yields? A: FFPE cross-linking fragments DNA and requires specialized reversal. Standard kits fail. Use a kit specifically designed for FFPE or ancient DNA that includes prolonged proteinase K digestion at high temperature (e.g., 56°C overnight) and a de-crosslinking step. A 2023 comparative analysis in Journal of Molecular Diagnostics found that an optimized FFPE-specific protocol recovered DNA fragments >1kb in length, which was critical for accurate taxonomic assignment of tumor-associated bacteria.
Experimental Protocols Cited
Protocol 1: Benchmarking Extraction Kits for Resistome Analysis (Stool)
- Homogenize 200 mg of stool in 1 mL lysis buffer.
- Split sample: Process aliquots with four different kits (e.g., QIAamp PowerFecal, MoBio Powersoil, bead-beating + phenol-chloroform, enzymatic lysis only).
- For bead-beating kits: Lysate is subjected to mechanical disruption (0.1 mm glass beads) on a homogenizer at 6.5 m/s for 45s.
- Complete extraction per manufacturer’s instructions.
- Quantify DNA yield (Qubit), assess fragment size (TapeStation).
- Perform shotgun metagenomic sequencing on equal mass inputs.
- Analyze reads against the Comprehensive Antibiotic Resistance Database (CARD).
Protocol 2: Optimized Extraction for Soil Actinobacteria
- Take 0.5 g of soil into a lysing matrix E tube.
- Add 800 µL of phosphate buffer (pH 8.0) and 200 µL of 10% SDS lysis buffer.
- Perform bead-beating: 6.0 m/s for 4 minutes using a reciprocating homogenizer.
- Incubate at 70°C for 20 minutes with gentle mixing every 5 minutes.
- Centrifuge, transfer supernatant.
- Proceed with phenol-chloroform-isoamyl alcohol purification.
- Precipitate DNA with isopropanol, wash with 70% ethanol, resuspend in TE buffer.
Protocol 3: Viral Enrichment from Sputum for Viromics
- Dilute 1 mL of sputum in 4 mL of SM Buffer. Vortex thoroughly.
- Centrifuge at 5,000 x g for 10 min at 4°C to remove cells/debris.
- Filter supernatant through a 0.22 µm PES filter.
- Treat filtrate with 50 U/mL Benzonase and 5 mM MgCl2. Incubate at 37°C for 1 hour to degrade free nucleic acids.
- Concentrate viral particles using 100kDa centrifugal filters.
- Lysate with Proteinase K and SDS at 56°C for 1h.
- Extract viral DNA using a standard column-based kit.
Data Presentation
Table 1: Impact of Extraction Method on Key Research Outcomes
| Study Focus (Sample) | Extraction Method A (Harsh) | Extraction Method B (Gentle) | Key Finding Influence |
|---|---|---|---|
| Antibiotic Resistome (Stool)Yield/Diversity | High yield (45 ± 12 ng/mg); High G+ recovery | Lower yield (22 ± 8 ng/mg); Lower G+ recovery | Method A revealed 33% more ARGs, identifying different primary resistance mechanisms. |
| CRC Biomarkers (Tissue)Fusobacterium spp. Abundance | 8.2% relative abundance | 2.1% relative abundance | Biomarker signature shifted from Fusobacterium-linked (Method A) to Prevotella-linked (Method B). |
| Soil Natural Products (Soil)Actinobacteria Read % | 25.4 ± 3.1% of bacterial reads | 6.8 ± 2.7% of bacterial reads | Method A enabled recovery of 15 novel biosynthetic gene clusters missed by Method B. |
| Lung Virome (Sputum)Host DNA Contamination | 15% host reads (with filtration+DNase) | >80% host reads (no filtration/DNase) | Method A increased viral sequencing depth 7-fold, enabling identification of novel temperate phages. |
Table 2: Research Reagent Solutions Toolkit
| Reagent / Material | Function in Metagenomic DNA Extraction |
|---|---|
| Lysing Matrix E Tubes | Contains ceramic/silica beads for mechanical disruption of tough cell walls (e.g., spores, Gram-positives). |
| Proteinase K | Broad-spectrum serine protease; degrades proteins and inactivates nucleases, crucial for complex samples. |
| Benzonase Nuclease | Degrades all forms of DNA and RNA; used in virome protocols to remove contaminating free host nucleic acids. |
| Inhibitor Removal Technology (IRT) Buffers | Contains proprietary compounds to bind and remove humic acids, polyphenols, and other PCR inhibitors from soil/plants. |
| Magnetic Beads (SPRI) | Size-selective binding of DNA fragments for cleanup and size selection; critical for building NGS libraries. |
| Phenol-Chloroform-Isoamyl Alcohol | Organic extraction removes proteins and lipids; used in rigorous protocols for maximum yield and purity. |
| Guanidine Thiocyanate (GuSCN) | Chaotropic agent that denatures proteins, inhibits RNases, and promotes nucleic acid binding to silica columns. |
Mandatory Visualizations
Title: Extraction Choice Drives Divergent Research Outcomes
Title: Viral Enrichment Workflow for Clean Metagenomes
Emerging Standards and Best Practice Guidelines from Consortia
Technical Support Center: Troubleshooting DNA Extraction for Metagenomics
This technical support resource, framed within the thesis "Optimizing DNA Extraction Methods for Complex Metagenomic Samples in Drug Discovery Pipelines," addresses common experimental challenges by aligning with emerging standards from consortia like the International Sequencing Standards Consortium (ISSC) and the Earth Microbiome Project (EMP).
FAQs & Troubleshooting Guides
Q1: My extracted DNA yield from a soil sample is consistently low. What are the best practice recommendations for improving biomass lysis? A: Low yield often indicates inefficient cell lysis. Current consortium guidelines (e.g., EMP) recommend a sequential, mechanical + chemical lysis approach.
- Troubleshooting Steps:
- Increase Mechanical Disruption: Use bead-beating with a mixture of zirconia/silica beads (0.1 mm and 0.5 mm) for 3-5 minutes at high speed. This is now a standard for robust lysis of Gram-positive bacteria and spores.
- Combine with Enzymatic Lysis: Follow bead-beating with an incubation step using a lysozyme (20 mg/ml, 37°C for 30 min) and proteinase K (20 mg/ml, 55°C for 60 min) cocktail.
- Validate with a Spike-in Control: Use an internal standard (e.g., a known quantity of Pseudomonas aeruginosa cells not native to your sample) to quantify and correct for lysis efficiency. Recovery of <80% suggests protocol optimization is needed.
Q2: I am detecting host (human) DNA contamination in my stool metagenome prep, impacting sequencing depth for microbial targets. How do I mitigate this? A: Host contamination is a critical issue in clinical metagenomics. Best practice guidelines from the Human Microbiome Project (HMP) and ISSC recommend selective lysis and/or post-extraction depletion.
- Troubleshooting Steps:
- Selective Lysis: Begin with a gentle enzymatic lysis step (lysozyme only, 37°C for 15 min) to lyse microbial cells, followed by centrifugation to pellet host cells. Transfer the supernatant containing microbial DNA for further processing.
- Propidium Monoazide (PMA) Treatment: Prior to lysis, treat samples with PMA (e.g., 50 µM for 10 min in the dark, then 15 min photo-activation). This dye penetrates compromised (microbial) cells and cross-links their DNA, inhibiting its amplification, while intact host cells may exclude it. Note: This requires validation for your sample type.
- Post-Extraction Depletion: Use commercial kits with probes targeting human mitochondrial and ribosomal DNA to deplete contaminating sequences after extraction.
Q3: My extracted DNA shows shearing/Fragment length is too short for long-read sequencing library prep. Which steps should I review? A: Excessive DNA shearing compromises assembly. Standards emphasize gentle handling and inhibitor removal.
- Troubleshooting Steps:
- Minimize Mechanical Shear: Avoid vigorous vortexing after cell lysis. Use wide-bore pipette tips for all DNA handling steps.
- Optimize Bead-Beating: Excessive bead-beating time is a common cause. Follow a time-course experiment (e.g., 30s, 60s, 90s, 120s) to find the minimum effective time for your sample matrix.
- Assess Inhibitor Removal: Polysaccharides and humic acids can co-precipitate with DNA, leading to forced aggressive pipetting. Ensure you are using validated inhibitor removal steps (e.g., CTAB precipitation, column wash buffers with guanidine thiocyanate).
Q4: How do I quantitatively compare the performance of different extraction kits for my specific sample type (e.g., biofilm)? A: Consortium standards require holistic assessment beyond just yield. Perform a standardized benchmarking experiment as per the table below.
Experimental Protocol: Kit Benchmarking
- Sample Homogenization: Aliquot the same sample (e.g., biofilm) into 5x 0.25 g replicates.
- Kit Application: Extract DNA using 3-4 different commercial kits and one manual gold-standard method (e.g., phenol-chloroform-isoamyl alcohol).
- Spike-in Control: Add a known amount of synthetic or cultured cell control (e.g., Bacillus subtilis spores) to each aliquot pre-extraction.
- QC Analysis: Quantify total DNA (Qubit), assess purity (A260/A280, A260/A230), and profile fragment size (Bioanalyzer).
- Downstream Analysis: Perform 16S rRNA gene amplicon sequencing and/or shallow shotgun sequencing on all extracts. Analyze for microbial community composition bias, alpha diversity, and spike-in recovery rate.
Table 1: Quantitative Benchmarking of Four DNA Extraction Methods from a Synthetic Biofilm Community
| Method | Avg. Yield (ng DNA/g) | A260/A280 | A260/A230 | Avg. Fragment Size (bp) | Spike-in Recovery (%) | Microbial Richness (Chao1 Index)* |
|---|---|---|---|---|---|---|
| Mechanical + Phenol-Chloroform | 5,200 | 1.82 | 2.05 | >23,000 | 95 | 148 ± 12 |
| Kit A (PowerSoil Pro) | 4,850 | 1.80 | 2.10 | ~10,000 | 89 | 145 ± 10 |
| Kit B (FastDNA Spin) | 5,500 | 1.75 | 1.85 | ~5,000 | 92 | 135 ± 15 |
| Kit C (DNeasy PowerBiofilm) | 4,100 | 1.84 | 2.15 | ~15,000 | 85 | 151 ± 8 |
*Based on 16S rRNA gene sequencing (V4 region). Higher Chao1 indicates better recovery of rare taxa.
Standardized Metagenomic DNA Extraction & QC Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function in Metagenomic DNA Extraction |
|---|---|
| Zirconia/Silica Beads (0.1, 0.5 mm mix) | Mechanically disrupts robust cell walls (Gram-positive, spores) and environmental aggregates during bead-beating. |
| Lysozyme & Proteinase K Enzyme Cocktail | Chemically degrades peptidoglycan (lysozyme) and proteins (proteinase K) for comprehensive enzymatic lysis. |
| Cetyltrimethylammonium Bromide (CTAB) | A cationic detergent effective in precipitating polysaccharides and humic acid inhibitors common in soil/plant samples. |
| Guanidine Thiocyanate (GuSCN) | A chaotropic salt used in lysis buffers to denature proteins and in wash buffers to promote DNA binding to silica. |
| Propidium Monoazide (PMA) | A DNA-intercalating dye used selectively before lysis to inhibit PCR amplification from membrane-compromised (dead) cells. |
| Solid Phase Reversible Immobilization (SPRI) Beads | Magnetic beads that selectively bind DNA by size for purification, concentration, and buffer exchange. |
| Internal Spike-in Control (e.g., gBlock, Cultured Cells) | A non-native, quantified DNA or cell standard added pre-extraction to benchmark and normalize for process efficiency. |
Conclusion
Successful metagenomic analysis is inextricably linked to the initial DNA extraction, a step that imposes the first and often most significant bias on the dataset. As outlined, the choice of method must be a deliberate, sample-informed decision balancing yield, integrity, and representativeness. Foundational understanding guides hypothesis, methodological rigor ensures reproducibility, troubleshooting safeguards data quality, and comparative validation aligns technique with translational intent. Future directions point towards increased standardization, the development of more robust universal protocols for low-biomass clinical samples, and integrated extraction-sequencing workflows that minimize bias. For biomedical and clinical research—particularly in drug development and microbiome-based diagnostics—investing in optimized, validated DNA extraction is not merely a preliminary step but a foundational investment in the accuracy, reliability, and ultimate impact of the research.